36 KiB
| title | date | updated | tags | categories | keywords | description | top_img | comments | cover | toc | toc_number | toc_style_simple | copyright | copyright_author | copyright_author_href | copyright_url | copyright_info | katex | highlight_shrink | aside |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Variational graph auto-encoders for miRNA-disease association prediction | 2020-11-26 17:32:15 | 2021-02-06 15:46:57 | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | true | <nil> | <nil> |
ABSTRACT
累积的实验研究已经证明,microRNAs(MiRNAs)在各种基本和重要的生物学过程中以及在许多复杂的人类疾病的发展中发挥着关键作用。 因此,探索miRNAs与疾病的关系,有助于理解miRNAs与疾病的机制,有助于复杂疾病的检测、诊断和治疗。由于通过传统的生物学实验来鉴定miRNA与疾病的相关性既耗时又昂贵,因此一种有效的计算预测方法是很有吸引力的。在这项研究中,我们提出了一种基于变化图自动编码器的miRNA-疾病关联预测深度学习框架(VGAE-MDA)。VGAE-MDA首先从miRNA-miRNA相似性、疾病-疾病相似性和已知miRNA-疾病关联构成的异构网络中获得miRNA和疾病的表示。然后,VGAE-MDA构建了两个子网络:基于miRNA的网络和基于疾病的网络。结合基于异构网络的表示法,部署了两个变分图自动编码器(VGAE),分别计算两个子网的miRNA-疾病关联分数。最后,VGAE-MDA通过整合来自这两个训练网络的分数来获得miRNA-疾病对的最终预测关联分数。与以往的模型不同,VGAE-MDA模型可以有效地抑制随机抽取负值样本所带来的噪声影响。此外,图卷积神经网络(GCN)的使用可以自然地融合图结构中的节点特征,而变分自动编码器(VAE)则从数据分布的角度利用潜在变量来预测关联。实验结果表明,VGAE-MDA在miRNA疾病关联预测方面优于目前最先进的方法。此外,通过案例分析进一步证明了该模型的有效性。
1. Introduction
[未翻译]
为了克服现有miRNA与疾病关联预测方法的局限性,我们提出了一种基于变化图的MDA深度学习框架(VGAEMDA)。据统计,图形卷积网络(GCN)已被广泛应用于关联预测中,如多视图卷积网络的链接预测[36],GCN关注网络的疾病相关RNA关联预测[37],以及GCN的miRNA与耐药性关联预测[38]。GCN可以直接对图形进行神经网络卷积运算,在图形的关联预测方面表现出很好的性能。此外,变分自动编码器(VAE)也被广泛应用于不同的领域,如药物反应预测[39]、文本分类[40]。VAE利用潜在变量从数据分布的角度进行预测,是一种生成模型。在我们的研究中,我们首先整合不同类型的信息构建了一个异构网络,该网络包括miRNA-miRNA相似网络(由miRNA功能相似、miRNA表达相似和高斯核相似构建)、疾病-疾病相似网络(由疾病语义相似和高斯核相似构建)和验证的miRNA-疾病关联网络。基于多个不同来源的相似度计算可以包含网络中更多的信息,有利于捕捉深层的交互。基于这种异构网络,我们得到了miRNA特征和疾病特征,这种表示特征比直接线性组合不同方面特征的表示特征更合理。然后,利用VAE擅长处理缺失数据的特性,通过给出不同阈值的相似度网络,构造了两个VGAE:基于miRNA的网络(不含疾病-疾病关联)和基于疾病的网络(不含miRNA-miRNA关联)。然后,基于第一步得到的miRNA和疾病特征,用两个VGAE计算每个miRNA-疾病对的两个预测分数。在VGAE的编码部分,GCN从网络结构和节点特征中学习输入数据的嵌入和分布,而在解码部分,VGAE重构原始输入数据并得到预测得分。最后,我们将两个预测分数进行整合,以获得每对miRNA疾病的最终预测分数。
2. Materials and methods
2.1. 已证实的人类miRNA疾病关联
已知的人类miRNA疾病关联是从实验验证的miRNA疾病数据库HMDD v2.0[41]和数据库miR2Disease[42]中获得的。我们将HMDD v2.0作为数据库D1,将不同阶段的miRNA组合在一起(如Has-let-7a-1和Has-let-7a-2),包括495个miRNA和383个疾病之间的5441对正关联。HMDD几乎包含了miR2疾病的所有数据。将HMDD v2.0与miR2 Disease结合后,我们得到了数据库D2,其中包含了577个miRNA和336种疾病之间的6313个实验验证的miRNA与疾病的关联。为了将我们的模型与以前基于相同数据库的方法进行比较,我们分别在D1和D2对我们的VGAE-MAD模型进行了评估。
2.2. MiRNA-miRNA similarity
2.3. Disease-disease similarity
2.4. MiRNA病异质网络和两个子网络
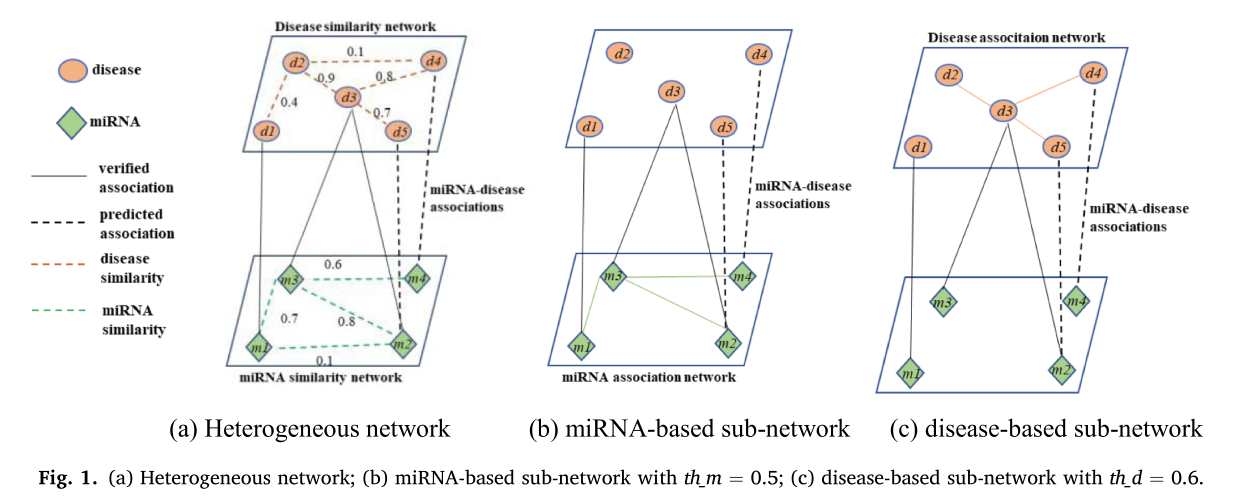
根据miRNA-miRNA相似网络Sm、疾病相似网络Sd和实验验证的miRNA疾病关联,我们构建了一个异质网络,如图1(A)所示。请注意,两个相似网络是边加权图。
 根据异质网络,我们可以得到每个miRNA和疾病的特征表示。异构网络的邻接矩阵H描述如下:
$H=\left[\begin{array}{ll}
S_{m} & A \
A^{T} & S_{d}
\end{array}\right]$
其中是矩阵$\mathrm{A}$的转置,设和$N_{d}$分别表示miRNAs的数量和疾病的数量。然后,矩阵$H$的前$N_{m}$行表示$N_{m}$ 个miRNA,矩阵$H$的最后$N_d$行表示$N_d$个疾病。也就是说,第i个miRNA的特征向量为$m_{i}=\left[S m_{i 1}, S m_{i 2}, \ldots, S m_{i N m}, A_{i 1}, A_{i 2}, \ldots, A_{i N d}\right]$,疾病的特征向量为$d_{j}=\left[A_{j 1}, A_{j 2}, \ldots, A_{j N d}, S d_{j 1}, S d_{j 2}, \ldots, S d_{j N d}\right]$。
接下来,我们将构建这两个子网。基于miRNA的子网络是在miRNA-miRNA相似网络和验证的miRNA-疾病关联网络的基础上构建的,但我们将边加权相似图二值化为具有相似阈值th_m的未加权网络,基于miRNA的子网络如图1(B)所示。同样,我们构造了基于疾病的子网络,如图1(C)所示,阈值为th_d。
根据异质网络,我们可以得到每个miRNA和疾病的特征表示。异构网络的邻接矩阵H描述如下:
$H=\left[\begin{array}{ll}
S_{m} & A \
A^{T} & S_{d}
\end{array}\right]$
其中是矩阵$\mathrm{A}$的转置,设和$N_{d}$分别表示miRNAs的数量和疾病的数量。然后,矩阵$H$的前$N_{m}$行表示$N_{m}$ 个miRNA,矩阵$H$的最后$N_d$行表示$N_d$个疾病。也就是说,第i个miRNA的特征向量为$m_{i}=\left[S m_{i 1}, S m_{i 2}, \ldots, S m_{i N m}, A_{i 1}, A_{i 2}, \ldots, A_{i N d}\right]$,疾病的特征向量为$d_{j}=\left[A_{j 1}, A_{j 2}, \ldots, A_{j N d}, S d_{j 1}, S d_{j 2}, \ldots, S d_{j N d}\right]$。
接下来,我们将构建这两个子网。基于miRNA的子网络是在miRNA-miRNA相似网络和验证的miRNA-疾病关联网络的基础上构建的,但我们将边加权相似图二值化为具有相似阈值th_m的未加权网络,基于miRNA的子网络如图1(B)所示。同样,我们构造了基于疾病的子网络,如图1(C)所示,阈值为th_d。
2.5. 图卷积网络
图卷积网络(GCN)[46,47]通常以图为输入,综合相邻节点的特征向量和图的结构信息来学习每个节点的特征向量。GCN既能有效地学习网络中节点的图拓扑结构,又能有效地学习网络中节点的属性信息。标准GCN及其变体可以显著提高许多网络相关预测任务的性能,如lncRNA-疾病关联预测[48]、用户-项目链接预测[38]和miRNA-耐药性预测[38]。
目前,根据图上局部卷积滤波器的定义,基于图的数据分析方法大致可以分为两类:基于空间的方法和基于谱的方法。基于空间的方法利用相邻节点的局部信息来构造滤波器的局部化,如Niepert的工作[49]。如Bruna的研究[50]所述,基于空间的方法在匹配本地邻居的机制上有很多局限性。与基于空间的方法相比,基于谱的方法是基于图的拉普拉斯谱[47]设计的。通过使用在谱域中实现的Kronecker增量,它在图上提供了定义良好的局部化算子。谱GCN在图学习方面具有优异的性能。近年来,许多研究试图揭示光谱GCN的内在和优势。 Atwood和Towsley指出,GCN实际上可以解释为一个图形扩散核[51]。Li等人声称,它有效的关键原因是谱图卷积是对称拉普拉斯平滑的一种特殊形式[52]。在本研究中,我们使用谱GCN分别从2.4节中构建的两个子网络中学习miRNA和疾病表示。
具体地说,以基于miRNA的子网络为例,设为包含$N_m$个miRNA和个疾病的基于miRNA的子网络$G_m$的邻接矩阵。设$X$是$G_m$的节点集上的标量特征。然后,我们通过$G_m$学习了miRNA和疾病的嵌入。$L_{m}=D_{m}^{-1 / 2} A_{m} D_{m}^{-1 / 2}$是$G_m$的对称规格化拉普拉斯矩阵,是具有对角线入口的对角矩阵。$L_m$可以被特征分解为$L_{m}=U_{m} \wedge_{m} U_{m}^{T}$,其中$U_{m}$是对应的特征向量矩阵,$\wedge_{m}=\operatorname{diag}\left(\lambda_{1}, \lambda_{2}, \ldots, \lambda_{N}\right)$是特征值矩阵,。然后,通过下面的图傅立叶变换(GFT):将步骤t-1处$G_m$上的图信号转换为新信号: $X^{(t)}=U_{m} \wedge_{m} U_{m}^{T} X^{(t-1)}$ (13)
在等式(13)中,$\wedge_{m}$是由$G_m$确定的图形信号滤波器。 通过$\mathrm{X}^{\prime(\mathrm{t}-1)}=U_{m}^{T} X^{(t-1)}$将图形信号变换到谱域。在应用滤波器之后,可以通过$\mathrm{U}_{\mathrm{m}} \mathrm{X}^{\prime(\mathrm{t}-1)}$将图形信号逆变换到图形域。
在等式(13)中通过对滤波器$\wedge_{m}$进行参数化,参数滤波器可以通过有监督学习的方式学习期望的图滤波器,而GFT可以动态地调整每个域对图信号变换的重要性。因此,滤波器变为$\theta\left(\wedge_{m}\right)=\operatorname{diag}\left(\theta_{1} \lambda_{1}, \theta_{2} \lambda_{2}\right. , \left.\ldots, \theta_{\mathrm{m}} \lambda_{\mathrm{m}}\right)$和公式(13)更改为以下方程式: $X^{(t)}=U_{m}\left(\theta \wedge_{m}\right) U_{m}^{T} X^{(t-1)}$ (14)
等式(14) 仍然面临两个限制: 第一个, 获取特征向量矩阵和特征值矩阵的过程对于大型网络来说是昂贵的; 第二个, 每个节点由一个标量特征表示, 不足以捕捉每个节点之间微妙而复杂的交互。
对于第一个限制,Kipf等人。用一阶切比雪夫近似解它[47]。具体地说$\widetilde{A}=A_{m}+I_{m}$表示$G_m$的自环邻接矩阵,$I_{m}$是单位矩阵。,然后,GFT可以重新表述为: (15) 其中是标量参数,它是的第一个条目。这样,我们就不再需要对$G_m$进行特征分解,同时也减小了滤波器参数的大小,从而加快了训练过程。
对于第二个限制,可以通过将标量图信号$\mathrm{X}$扩展为矢量信号$\mathrm{X}{1} \in \mathrm{R}^{\mathrm{N}^{*} \mathrm{fm}}$来解决,同时,可以将矢量滤波器参数θ扩展为具有$F$个滤波器和输入通道的滤波器参数矩阵$\Theta \in R^{f m & F}$。最后,频谱GCN可以表示如下: $X^{(t)}=\widetilde{D}{m}^{-1 / 2} \widetilde{A}{m} \widetilde{D}{m}^{-1 / 2} X^{(t-1)} \Theta_{m}^{t-1}$ (16)
在我们的研究中,$\widetilde{A}$是基于miRNA的子网络$G_m$的邻接矩阵加上一个单位矩阵,$X^0$是从异构网络$H$获得的子网络$G_m$中所有节点的特征矩阵。
公式(16)是前馈神经网络的线性层,采用非线性激活函数 f() 来增强模型的表达能力。在我们的研究中,我们使用RELU激活函数,那么t层的特征提取可以定义如下: $X^{(t)}=\operatorname{relu}\left(\widetilde{D}{m}^{-\frac{1}{2}} \widetilde{A}{m} \widetilde{D}{m}^{-\frac{1}{2}} X^{(t-1)} \Theta{m}^{t-1}\right)$ 这样,我们就可以通过GCN得到基于miRNA的子网中每个节点的表示。相似的情况下,我们还可以得到基于疾病的子网络中每个节点的表示。
2.6. Variational graph auto-encoder
变化图自动编码器(VGAE)是在变分自动编码器[53,54]的基础上学习图结构数据的框架。该模型可以通过GCN自然地包含节点特征,然后利用潜在变量从数据分布的角度学习无向图的可解释的潜在表示[54]。整个VAE模型包括编码部分和解码部分。在编码部分,它以邻接矩阵A和特征矩阵X为输入,得到潜在变量z作为输出;在解码部分,VGAE根据潜在变量z得到重构的邻接矩阵A。
我们仍然以基于miRNA的子网络$G_m$为例,是$G_m$中的节点数,$A_m$是邻接矩阵,是$A_m$的度矩阵。$X$为特征矩阵,并进一步引入随机潜变量。接下来,我们从编码、解码和损失函数三个方面介绍了GCN。
编码器:首先,VGAE包括两层GCN。第一GCN层生成低维特征矩阵。其定义如下: $\begin{array}{l} \bar{X}=G C N\left(X, A_{m}\right)=\operatorname{ReLU}\left(\widetilde{A}{m} X W{0}\right)\ \widetilde{A}{m}=D^{-\frac{1}{2}}\left(A{m}+I\right) D^{-\frac{1}{2}} \end{array}$
第二个GCN层按如下方式生成数据分布: $\begin{array}{l} \mu \backslash ;=\backslash ; \operatorname{GCN} \mu\left(\mathrm{X}, \backslash ; A_{m}\right) \backslash ;=\widetilde{A}{m} \bar{X} W{\mu} \ \log \sigma=G C N \sigma\left(X, A_{m}\right)=\widetilde{A}{m} \bar{X} W{\sigma} \end{array}$
然后,我们可以得到潜在变量z,如下所示: $\begin{array}{l} \mathrm{z}=\mu+\sigma^{*} \varepsilon\ \text { where } \varepsilon \text { follows } N(0,1) \end{array}$
编码器也可以表示为: $q\left(z_{i} \mid X, A_{m}\right)=N\left(z_{i} \mid \mu_{i}, \operatorname{diag}\left(\sigma_{i}^{2}\right)\right)$
解码器:解码器由潜在变量$z$之间的内积定义,输出为重构的邻接矩阵,如下所示:
其中S是Logistic Sigmoid函数。 解码器可以表示为: $p\left(A_{m i j}=1 \mid z_{i}, z_{j}\right)=S\left(z_{i}^{T} z_{j}\right)$
损失函数:损失函数包括两部分,第一部分是目标$A_m$和输出之间的二元交叉熵,第二部分是和$p(Z)$之间的KL散度。损失函数定义如下: $L=E_{q\left(Z \mid X, A_{m}\right)}\left[\log p\left(A_{m} \mid Z\right)\right]-K L\left[q\left(Z \mid X, A_{m}\right) | p(Z)\right]$
2.7. VGAE-MDA用于miRNA与疾病关联的预测
在这一部分中,我们开发了一种基于VGAE的miRNA疾病预测方法VGAE-MDA。VGAE-MDA的框架如图2所示,包括四个步骤。
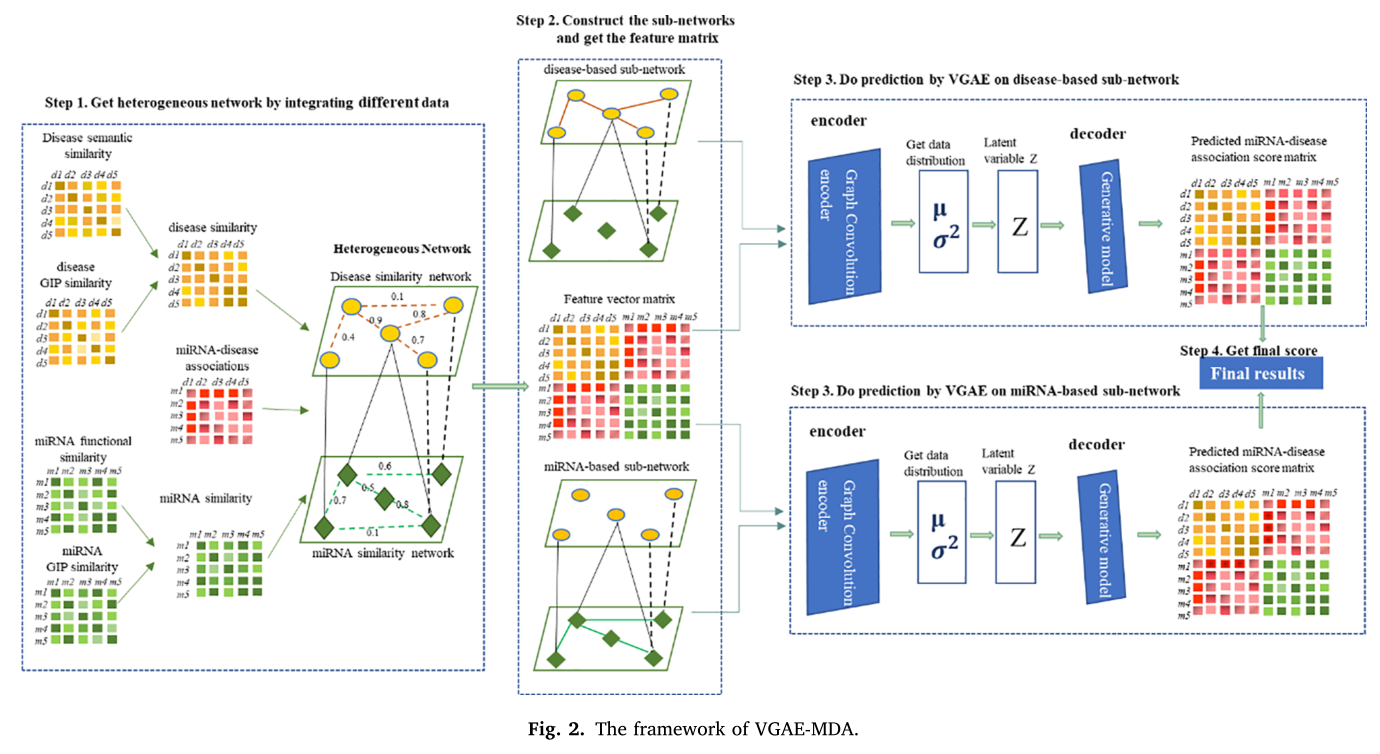 第一步,我们根据第2.2节得到的miRNA-miRNA相似度和第2.3节得到的疾病-疾病相似度构建了一个异构网络H,并验证了miRNA-疾病相关性。
根据这种异构网络,我们可以定义存储在特征矩阵X中的每个节点的特征。这种特征表示从不同的角度合理地描述了miRNA和疾病,在我们的研究中,它集成了高斯相互作用轮廓、miRNA功能相似性、疾病语义相似性以及miRNA与疾病的关联性。
第一步,我们根据第2.2节得到的miRNA-miRNA相似度和第2.3节得到的疾病-疾病相似度构建了一个异构网络H,并验证了miRNA-疾病相关性。
根据这种异构网络,我们可以定义存储在特征矩阵X中的每个节点的特征。这种特征表示从不同的角度合理地描述了miRNA和疾病,在我们的研究中,它集成了高斯相互作用轮廓、miRNA功能相似性、疾病语义相似性以及miRNA与疾病的关联性。
第二步,我们根据2.4节构造了两个具有两个不同相似度阈值TH_m和TH_d的子网络,在本研究中,加权相似网络被二值化为未加权网络。此外,由于VGAE是一种产生式模型,在处理数据丢失问题方面具有很大的优势,因此我们从异构网络中创建了两个子网。实验结果还表明,两个子网络比只有一个异构网络具有更好的性能。
步骤3,VGAE分别用这两个子网络进行预测,为每个miRNA-疾病对计算两个预测得分Score_m和Score_d。GCN可以自然地合并每个节点邻居的特征,而VAE可以根据数据分布执行预测。
步骤4,通过基于miRNA的分数_m和基于疾病的分数_d的平均值得到最终的关联分数。
3. Results and discussion
3.1. Performance evaluation
在实验中,采用10倍交叉验证(10-CV)、5倍交叉验证(5-CV)和局部5-CV来评估VGAE-MDA的预测性能。对于5-CV和10F CV,我们将已知的正关联随机分为5折或10折,每折轮流作为测试样本,其余作为每次训练集。在局部5-CV中,对于一个特定的疾病d(I),我们将与d(I)的所有关联分成五层,每一层依次作为测试样本,而网络中的所有其他关联都被视为训练样本。在每次训练之前,当我们确定测试数据时,将miRNA-疾病关联矩阵中对应的所有标签都设置为0。
为了从不同方面评价预测结果,本研究使用了受试者工作特征(ROC)曲线下面积(AUC)、准确率和召回率下面积(AUPR)和F1-Score。根据不同评分阈值下的真阳性率(TPR)和假阳性率(FPR)绘制ROC曲线,其中TPR为正确识别的阳性样本与真实阳性样本的比例,假阳性率为被确认为阳性的阴性样本占所有阴性样本的百分比。Aupr曲线绘制了不同分数阈值下的查准率与召回率之间的关系,其中查准率是正确预测的样本与总样本的比率,召回率与TPR相同。F1分数是精确度和查全率的调和平均数。为了计算FPR,我们随机选择与负样本相同数量的未知关联。
3.2. Hyperparameters
在本研究中,几个超参数影响预测的性能。VGAE-MDA包括两个子VGAE模型,我们保持这两个模型的结构不变。优化器是ADAM优化器,第一隐层中的活动函数是RELU[54],而第二隐层是线性活动函数。根据先前的研究,学习率(LR)被设置为0.01。根据先前的研究[54],隐藏层1(H1)和隐藏层2(H2)中的单元数分别被设置为48和16。我们还尝试了更多的GCN隐含层,但是,当隐含层数超过2时,预测性能会下降。由于模型经过700次迭代训练后变得稳定,因此将Epochs(EP)设置为700。
 相似度得分阈值THm和THd对预测结果影响很大,因为它们决定了有多少miRNA-miRNA关联或疾病-疾病关联参与了训练。
我们将阈值th_m和th_d设置为不同的值{0.1,0.2,…。,0.9,1}和图3显示了不同的Th_m或Th_d对数据库D1上5CV下的miRNA子网络或疾病子网络的预测性能的影响。显然,当Th_m选择为0.9时,基于miRNA的VGAE的性能最好,当Th_d选择为0.9时,基于疾病的VGAE的性能最好。我们假设当两个子VGAE模型都获得最好的性能时,最终的预测结果是最好的。图3还表明,参数Th_m对基于miRNA的VGAE性能的影响大于参数Th_d对基于疾病的VGAE性能的影响。
相似度得分阈值THm和THd对预测结果影响很大,因为它们决定了有多少miRNA-miRNA关联或疾病-疾病关联参与了训练。
我们将阈值th_m和th_d设置为不同的值{0.1,0.2,…。,0.9,1}和图3显示了不同的Th_m或Th_d对数据库D1上5CV下的miRNA子网络或疾病子网络的预测性能的影响。显然,当Th_m选择为0.9时,基于miRNA的VGAE的性能最好,当Th_d选择为0.9时,基于疾病的VGAE的性能最好。我们假设当两个子VGAE模型都获得最好的性能时,最终的预测结果是最好的。图3还表明,参数Th_m对基于miRNA的VGAE性能的影响大于参数Th_d对基于疾病的VGAE性能的影响。
3.3. 影响预测性能的因素
在本节中,我们将分析对VGAE-MDA性能有重要影响的组件。其中一个组件是GCN编码器,另一个是两个子网。
3.3.1. Effect of GCN encoders
GCN可以在图的层次上自然地融合节点及其邻居的特征,有助于发现网络中的深层交互。通过比较VGAE-MDA的预测性能和使用常规变分自动编码器的VAE-MDA模型的性能,研究了GCN编码器的性能。在相同的参数设置下,我们分别在数据库D1和D2上训练VGAE-MDA和VAE-MDA,每个模型运行20次。然后,5-CV和10-CV的预测结果的平均值如图4所示。
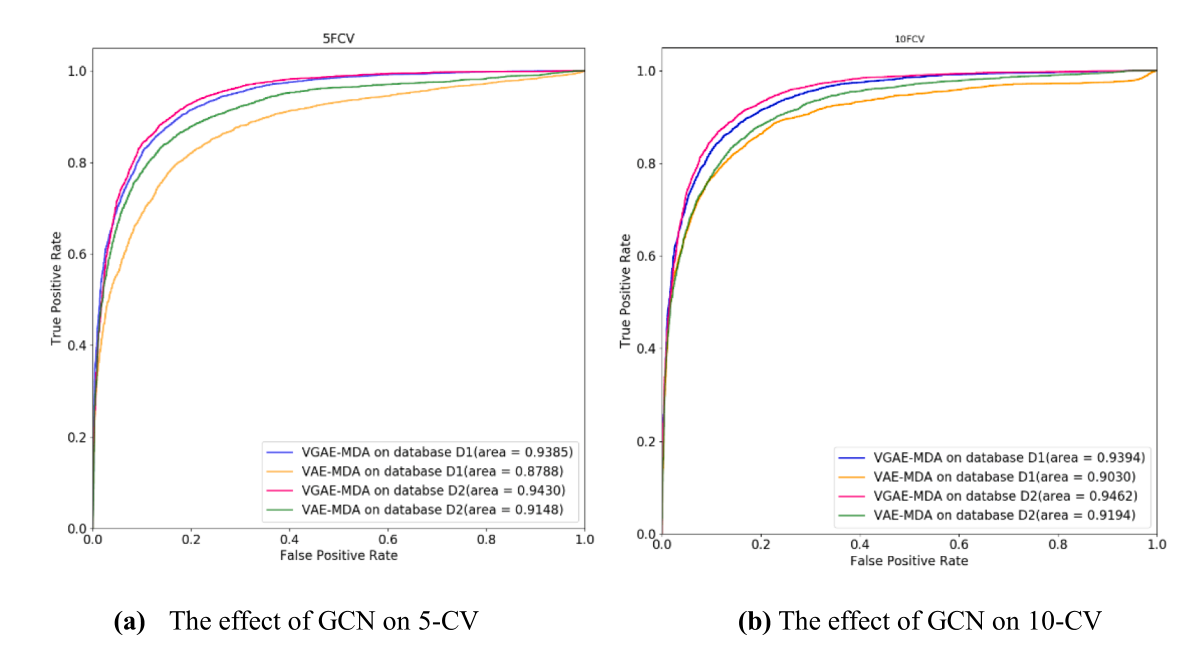 图4表明,在数据库D1和D2上,VGAE-MDA在5-CV和10-CV方面都比VAEMDA有更好的性能,这意味着GCN对VGAE-MDA的预测性能有很大的贡献。同样的方法,在数据集D2上的结果优于在数据集D1上的结果,使用10-CV的结果略好于使用5-CV的结果,这表明训练数据越多,模型的性能越好。
图4表明,在数据库D1和D2上,VGAE-MDA在5-CV和10-CV方面都比VAEMDA有更好的性能,这意味着GCN对VGAE-MDA的预测性能有很大的贡献。同样的方法,在数据集D2上的结果优于在数据集D1上的结果,使用10-CV的结果略好于使用5-CV的结果,这表明训练数据越多,模型的性能越好。
3.3.2 两个子网络的影响
在本研究中,我们从异构网络中创建了两个子网络。我们通过对基于疾病的子网络(Disease-VGAE)、基于miRNA的子网络(miRNA-VGAE)、异构网络(H-VGAE)和VGAE-MDA的预测结果的比较,证明了这一优势。
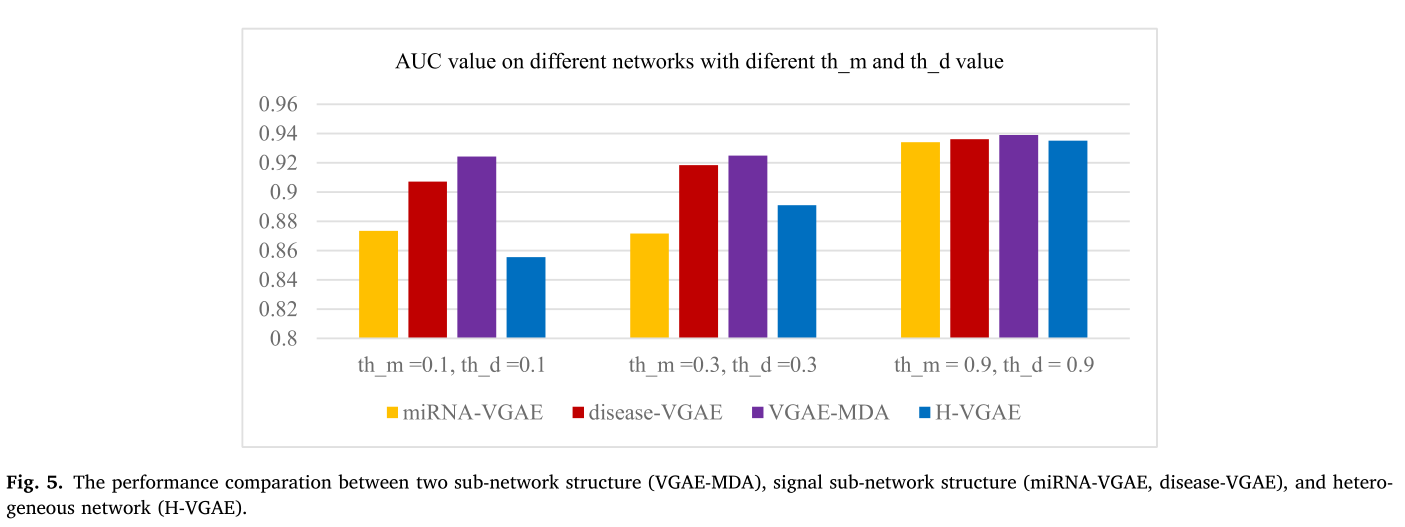 图5显示了三组不同参数th_m和th_d的比较结果,不同的th_m和th_d值对应于不同的miRNA-miRNA网络和疾病-疾病网络。在三组不同参数下的仿真结果表明,两个子网络结构(VGAE-MDA)的性能总是最好的,说明这两个子网络结构也对VGAE-MDA的良好性能有贡献。
图5显示了三组不同参数th_m和th_d的比较结果,不同的th_m和th_d值对应于不同的miRNA-miRNA网络和疾病-疾病网络。在三组不同参数下的仿真结果表明,两个子网络结构(VGAE-MDA)的性能总是最好的,说明这两个子网络结构也对VGAE-MDA的良好性能有贡献。
3.4. 与近几年已有的工作相比
已经提出了许多miRNA与疾病关联的预测方法。然而,现有方法中的数据集或评价方法并不一致。为了将VGAEMDA与现有的方法进行公平的比较,我们从以下几个方面进行了实验。
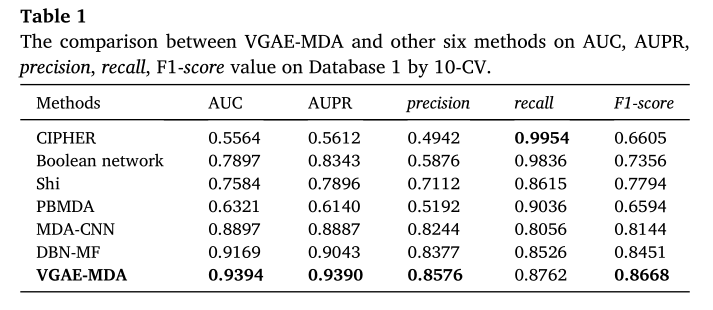 首先,我们在同一数据库D1上用10-CV从不同的角度比较了VGAE-MDA与六种竞争方法的预测结果。这六种相互竞争的方法包括基于网络的回归模型(密码)[55]、基于布尔网络的方法(布尔)[21]、基于随机游走的相似性方法(SHI)[27]、基于路径的方法(PBMDA)[56]、基于机器学习的方法(MDA-CNN)[57]和基于深度学习的方法(DBNMF)[58]。比较结果如表1所示。很明显,VGAE-MDA在AUC值、AUPR值、精度和F1得分方面表现最好。尽管VGAE在召回中得不到最高值,但它的F1得分是最高的,比召回更平衡。综上所述,VGAE-MDA的预测性能优于其他竞争方法。
首先,我们在同一数据库D1上用10-CV从不同的角度比较了VGAE-MDA与六种竞争方法的预测结果。这六种相互竞争的方法包括基于网络的回归模型(密码)[55]、基于布尔网络的方法(布尔)[21]、基于随机游走的相似性方法(SHI)[27]、基于路径的方法(PBMDA)[56]、基于机器学习的方法(MDA-CNN)[57]和基于深度学习的方法(DBNMF)[58]。比较结果如表1所示。很明显,VGAE-MDA在AUC值、AUPR值、精度和F1得分方面表现最好。尽管VGAE在召回中得不到最高值,但它的F1得分是最高的,比召回更平衡。综上所述,VGAE-MDA的预测性能优于其他竞争方法。
其次,我们在数据库D1上将VGAE-MDA与全局5-CV中的其他10种方法进行了比较。比较的10种方法包括NIMCGCN[59]、BNPMDA[60]、ABMDA[61]、EDTMDA[62]、MDHGI[63]、LRSSLMDA[32]、PBMDA[56]、mcmda[31]、MaxFlow[64]和HDMP[22]。
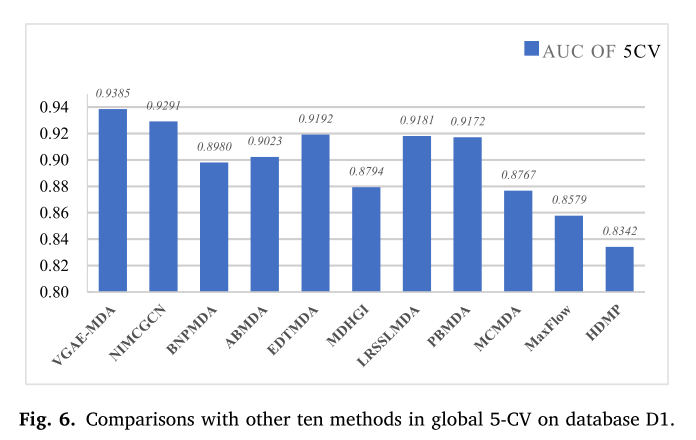 图6显示了5-CV的所有AUC值。对于VGAEMDA,进行了20个5-CV以减小5-CV样本分割的影响,平均AUC值为0.9385,明显大于比较方法。
图6显示了5-CV的所有AUC值。对于VGAEMDA,进行了20个5-CV以减小5-CV样本分割的影响,平均AUC值为0.9385,明显大于比较方法。
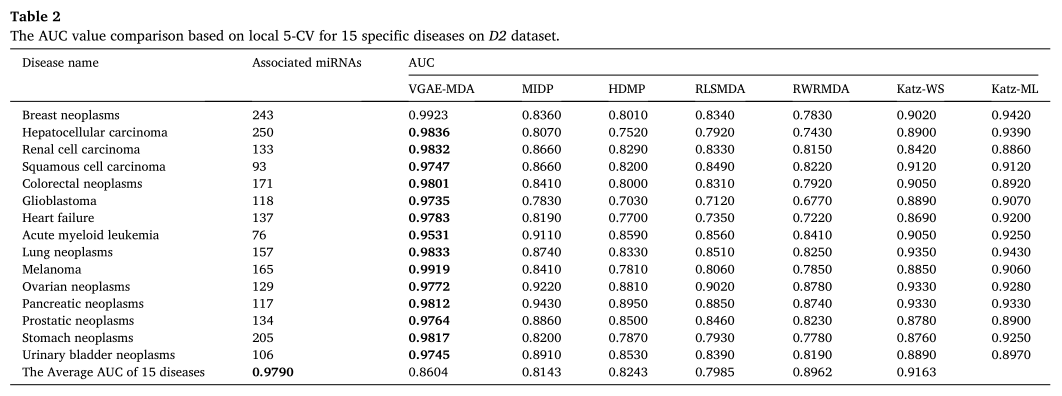 最后,一些研究提供了数据集D2上15种特殊疾病的局部5-CV结果,例如MIDP、HDMP、RLSMDA、RWRMDA、KatzWS和Katz-ML。在这一部分中,我们还对相同的15种特定疾病在相同的数据集D2上进行了实验,以比较它们的性能。结果如表2所示。显然,VGAEMDA在所有疾病和平均结果上都取得了最好的性能。平均AUC值为0.979,大大超过了其他预测方法。
最后,一些研究提供了数据集D2上15种特殊疾病的局部5-CV结果,例如MIDP、HDMP、RLSMDA、RWRMDA、KatzWS和Katz-ML。在这一部分中,我们还对相同的15种特定疾病在相同的数据集D2上进行了实验,以比较它们的性能。结果如表2所示。显然,VGAEMDA在所有疾病和平均结果上都取得了最好的性能。平均AUC值为0.979,大大超过了其他预测方法。
3.5.案例研究
为了进一步证明VGAE-MDA在识别新的miRNA疾病关联方面的预测能力,VGAE-MDA在数据库D1上进行,用于预测所有未知的miRNA疾病关联,我们通过对疾病结肠癌的前50个候选miRNA进行案例研究。我们通过搜索文献和另外两个miRNA疾病关联数据库,即dbDEMC v2.0[65]和miRCancer[66]来验证这些预测的关联。DbDEMC数据库收集不同人类癌症中差异表达的miRNAs,dbDEMC v2.0添加了更多从表达数据中获得的与癌症相关的miRNAs。MiRCancer是一个在自动提取后手动确认的数据库。 我们得到特定疾病与所有miRNA之间的所有关联得分,然后从原始miRNA-疾病关联矩阵中剔除值等于1的miRNA。在此之后,所有剩余的miRNA都按照预测分数从大到小的顺序进行排序。
表3显示了预测的与结肠癌相关的前50个miRNA。在dbDEMC数据库中,与结肠癌相关的miRNA有49204个,而在miRCancer数据库中与结肠癌相关的miRNA有756miRNA。根据这两个数据库和文献检索,在预测的50个miRNAs中有49个得到验证,只有1个miRNA Has-let-16没有得到确认,这意味着对新的结肠癌相关miRNAs的预测准确率为98%。

4 结论
MiRNAs已被证实与复杂的人类疾病有关。鉴定miRNA与疾病的相关性可以提高对疾病发病机制的认识,有助于疾病的治疗。随着越来越多的miRNA相关和疾病相关数据库在生物学实验的基础上被建立起来,研究人员开始关注通过计算方法预测新的miRNA与疾病的关联。在本研究中,针对以往研究的局限性,我们提出了一种基于变化图自动编码器的miRNA疾病关联预测深度学习框架。我们在两个不同的数据库D1和D2上进行了VGAE-MDA实验,实验结果表明,VGAE-MDA在不同的评价指标下取得了比以往的方法更好的预测性能。在数据库D1上,5-CV的AUC值为0.9385,10-CV的AUC值为0.9394。在D2数据库上,5-CV和10-CV的AUC值分别为0.9430和0.9462。此外,对于数据库D2上的本地5-CV,对于特定的15种疾病,其平均AUC值为0.979。
VGAE-MDA的优异性能归因于几个重要因素。首先,GCN的使用可以自然地整合邻居节点的信息,得到网络上每个节点的更好的表示,这有助于提取miRNAs与疾病之间的深层互动。其次,VAE是一种产生式模型,有助于处理缺失数据,因此我们创建了两个子网络。第3.3.2节的实验结果表明,该策略是有益的。最后,不同类型数据库的集成也有利于预测新的miRNA-疾病关联,从异构网络中得到的miRNA和疾病的表示比简单地将不同层次的特征线性组合得到的miRNA和疾病的表示更合理。
虽然VGAE-MDA在预测新的miRNA疾病关联方面表现出很好的性能,但也有一些局限性。例如,该框架包含两个深度学习网络,算法复杂度相对较高,当数据库变得很大时就不友好了。此外,相似度的计算还需要进一步改进。今后,我们将尝试用不同的相似度计算方法将更多不同的数据库集成到相似度网络中。
References
[1] D.P. Bartel, MicroRNAs: genomics, biogenesis, mechanism, and function, Cell 116 (2) (2004) 281–297. [2] V. Ambros, MicroRNA pathways in flies and worms: growth, death, fat, stress, and timing, Cell 113 (6) (2003) 673–676. [3] X. Karp, V. Ambros, Encountering microRNAs in cell fate signaling, Science 310 (5752) (2005) 1288–1289. [4] E.A. Miska, How microRNAs control cell division, differentiation and death, Curr. Opin. Genet. Dev. 15 (5) (2005) 563–568. [5] K.D. Taganov, M.P. Boldin, K.-J. Chang, D. Baltimore, NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses, Proc. Natl. Acad. Sci. 103 (33) (2006) 12481–12486. [6] F. Meng, R. Henson, H. Wehbe–Janek, K. Ghoshal, S.T. Jacob, T. Patel, MicroRNA- 21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer, Gastroenterology 133 (2) (2007) 647–658. [7] M. Carleton, M.A. Cleary, P.S. Linsley, MicroRNAs and cell cycle regulation, Cell Cycle 6 (17) (2007) 2127–2132. [8] Y.-H. Taguchi, Inference of target gene regulation via miRNAs during cell senescence by using the MiRaGE server, Springer, Int. Conf. Intell. Comput. (2012) 441–446. [9] S. Griffiths-Jones, miRBase: microRNA sequences, targets and gene nomenclature, Nucl. Acids Res. 34 (90001) (2006) D140–D144, https://doi.org/10.1093/nar/ gkj112. [10] S. Hua, W. Yun, Z. Zhiqiang, Q. Zou, A discussion of micrornas in cancers, Curr. Bioinform. 9 (5) (2014) 453–462. [11] D. Madhavan, M. Zucknick, M. Wallwiener, K. Cuk, C. Modugno, M. Scharpff, S. Schott, J. Heil, A. Turchinovich, R. Yang, Circulating miRNAs as surrogate markers for circulating tumor cells and prognostic markers in metastatic breast cancer, Clin. Cancer Res. 18 (21) (2012) 5972–5982. [12] N. Kliese, P. Gobrecht, D. Pachow, N. Andrae, A. Wilisch-Neumann, E. Kirches, M. Riek-Burchardt, F. Angenstein, G. Reifenberger, M.J. Riemenschneider, miRNA- 145 is downregulated in atypical and anaplastic meningiomas and negatively regulates motility and proliferation of meningioma cells, Oncogene 32 (39) (2013) 4712. [13] S. Zhao, G. Yang, Y. Mu, D. Han, C. Shi, X. Chen, Y. Deng, D. Zhang, L. Wang, Y. Liu, MiR-106a is an independent prognostic marker in patients with glioblastoma, Neuro-oncology 15 (6) (2013) 707–717. [14] N. Lynam-Lennon, S.G. Maher, J.V. Reynolds, The roles of microRNA in cancer and apoptosis, Biol. Rev. 84 (1) (2009) 55–71. [15] Z. Huang, J. Shi, Y. Gao, C. Cui, S. Zhang, J. Li, Y. Zhou, Q. Cui, HMDD v3. 0: a database for experimentally supported human microRNA–disease associations, Nucleic Acids Res. 47 (D1) (2018) D1013–D1017. [16] A. Hamosh, Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders, Nucleic Acids Res. 33 (Database issue) (2004) D514–D517, https://doi.org/10.1093/nar/gki033. [17] X.-M. Zhao, K.-Q. Liu, G. Zhu, F. He, B. Duval, J.-M. Richer, D.-S. Huang, C.- J. Jiang, J.-K. Hao, L. Chen, Identifying cancer-related microRNAs based on gene expression data, Bioinformatics 31 (8) (2015) 1226–1234. [18] T. Keshava Prasad, R. Goel, K. Kandasamy, S. Keerthikumar, S. Kumar, S. Mathivanan, D. Telikicherla, R. Raju, B. Shafreen, A. Venugopal, Human protein reference database—2009 update, Nucl. Acids Res. 37 (suppl_1) (2008) D767–D772. [19] G.-M. Qin, R.-Y. Li, X.-M. Zhao, Identifying disease associated miRNAs based on protein domains, IEEE/ACM Transf. Comput. Biol. Bioinf. 13 (6) (2016) 1027–1035. [20] X. Zeng, X. Zhang, Q. Zou, Integrative approaches for predicting microRNA function and prioritizing disease-related microRNA using biological interaction networks, Briefings Bioinf. 17 (2) (2016) 193–203. [21] Q. Jiang, Y. Hao, G. Wang, L. Juan, T. Zhang, M. Teng, Y. Liu, Y. Wang, Prioritization of disease microRNAs through a human phenome-microRNAome network, BMC Syst. Biol. 4 (1) (2010) S2. [22] P. Xuan, K. Han, M. Guo, Y. Guo, J. Li, J. Ding, Y. Liu, Q. Dai, J. Li, Z. Teng, Prediction of microRNAs associated with human diseases based on weighted k most similar neighbors, PLoS One 8 (8) (2013), e70204. [23] S. Mørk, S. Pletscher-Frankild, A. Palleja Caro, J. Gorodkin, L.J. Jensen, Protein- driven inference of miRNA–disease associations, Bioinformatics 30 (3) (2014) 392–397. [24] G. Li, J. Luo, Q. Xiao, C. Liang, P. Ding, Predicting microRNA-disease associations using label propagation based on linear neighborhood similarity, J. Biomed. Inform. 82 (2018) 169–177. [25] M. Chen, Y. Zhang, A. Li, Z. Li, W. Liu, Z. Chen, Bipartite Heterogeneous Network Method Based on Co-neighbour for MiRNA–Disease Association Prediction, Front. Genet. 10 (2019) 385. [26] X. Chen, M.-X. Liu, G.-Y. Yan, RWRMDA: predicting novel human microRNA–disease associations, Mol. BioSyst. 8 (10) (2012) 2792–2798. [27] H. Shi, J. Xu, G. Zhang, L. Xu, C. Li, L. Wang, Z. Zhao, W. Jiang, Z. Guo, X. Li, Walking the interactome to identify human miRNA-disease associations through the functional link between miRNA targets and disease genes, BMC Syst. Biol. 7 (1) (2013) 101. [28] P. Xuan, K. Han, Y. Guo, J. Li, X. Li, Y. Zhong, Z. Zhang, J. Ding, Prediction of potential disease-associated microRNAs based on random walk, Bioinformatics 31 (11) (2015) 1805–1815. [29] X. Chen, G.-Y. Yan, Semi-supervised learning for potential human microRNA- disease associations inference, Sci. Rep. 4 (2014) 5501. [30] J. Luo, P. Ding, C. Liang, B. Cao, X. Chen, Collective prediction of disease- associated miRNAs based on transduction learning, IEEE/ACM Trans. Comput. Biol. Bioinf. 14 (6) (2016) 1468–1475. [31] J.-Q. Li, Z.-H. Rong, X. Chen, G.-Y. Yan, Z.-H. You, MCMDA: Matrix completion for MiRNA-disease association prediction, Oncotarget 8 (13) (2017) 21187. [32] X. Chen, L. Huang, LRSSLMDA: Laplacian regularized sparse subspace learning for MiRNA-disease association prediction, PLoS Comput. Biol. 13 (12) (2017), e1005912. [33] Y.-W. Niu, G.-H. Wang, G.-Y. Yan, X. Chen, Integrating random walk and binary regression to identify novel miRNA-disease association, BMC Bioinf. 20 (1) (2019) 59. [34] P. Xuan, H. Sun, X. Wang, T. Zhang, S. Pan, Inferring the disease-associated miRNAs based on network representation learning and convolutional neural networks, Int. J. Mol. Sci. 20 (15) (2019) 3648. [35] J. Luo, Q. Xiao, A novel approach for predicting microRNA-disease associations by unbalanced bi-random walk on heterogeneous network, J. Biomed. Inform. 66 (2017) 194–203. [36] Z. Li, Z. Liu, J. Huang, G. Tang, Y. Duan, Z. Zhang, Y. Yang, MV-GCN: multi-view graph convolutional networks for link prediction, IEEE Access 7 (2019) 176317–176328. [37] J. Zhang, X. Hu, Z. Jiang, B. Song, W. Quan, Z. Chen, Predicting Disease-related RNA Associations based on Graph Convolutional Attention Network, in: 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), IEEE, 2019, pp. 177–182. [38] Y.-A. Huang, P. Hu, K.C. Chan, Z.-H. You, Graph convolution for predicting associations between miRNA and drug resistance, Bioinformatics 36 (3) (2020) 851–858. [39] L. Ramp´aˇsek, D. Hidru, P. Smirnov, B. Haibe-Kains, A. Goldenberg, VAE: improving drug response prediction via modeling of drug perturbation effects, Bioinformatics 35 (19) (2019) 3743–3751. [40] S. Rashid, S. Shah, Z. Bar-Joseph, R. Pandya, Dhaka: variational autoencoder for unmasking tumor heterogeneity from single cell genomic data, bioRxiv (2018) 183863. [41] Y. Li, C. Qiu, J. Tu, B. Geng, J. Yang, T. Jiang, Q. Cui, HMDD v2. 0: a database for experimentally supported human microRNA and disease associations, Nucleic Acids Res. 42 (D1) (2013) D1070–D1074. [42] Q. Jiang, Y. Wang, Y. Hao, L. Juan, M. Teng, X. Zhang, M. Li, G. Wang, Y. Liu, miR2Disease: a manually curated database for microRNA deregulation in human disease, Nucleic Acids Res. 37 (Database) (2009) D98–D104, https://doi.org/ 10.1093/nar/gkn714. [43] D. Wang, J. Wang, M. Lu, F. Song, Q. Cui, Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases, Bioinformatics 26 (13) (2010) 1644–1650. [44] S. Bandyopadhyay, R. Mitra, U. Maulik, M.Q. Zhang, Development of the human cancer microRNA network, Silence 1 (1) (2010) 6. [45] X. Chen, G.-Y. Yan, Novel human lncRNA–disease association inference based on lncRNA expression profiles, Bioinformatics 29 (20) (2013) 2617–2624. [46] M. Defferrard, X. Bresson, P. Vandergheynst, Convolutional neural networks on graphs with fast localized spectral filtering, Advances in neural information processing systems (2016) 3844–3852. [47] T.N. Kipf, M. Welling, Semi-supervised classification with graph convolutional networks, arXiv preprint arXiv: 1609.02907 (2016). [48] P. Xuan, S. Pan, T. Zhang, Y. Liu, H. Sun, Graph Convolutional Network and Convolutional Neural Network Based Method for Predicting lncRNA-Disease Associations, Cells 8 (9) (2019) 1012. [49] M. Niepert, M. Ahmed, K. Kutzkov, Learning convolutional neural networks for graphs, Int. Conf. Mach. Learn. (2016) 2014–2023. [50] J. Bruna, W. Zaremba, A. Szlam, Y. LeCun, Spectral networks and locally connected networks on graphs, arXiv preprint arXiv:1312.6203 (2013). [51] J. Atwood, D. Towsley, Diffusion-convolutional neural networks, Adv. Neural Inf. Process. Syst. (2016) 1993–2001. [52] Q. Li, Z. Han, X.-M. Wu, Deeper insights into graph convolutional networks for semi-supervised learning, Thirty-Second AAAI Conference on Artificial Intelligence, 2018. [53] D.P. Kingma, M. Welling, Stochastic gradient VB and the variational auto-encoder. Second International Conference on Learning Representations, ICLR, 2014. [54] T.N. Kipf, M. Welling, Variational graph auto-encoders, arXiv preprint arXiv: 1611.07308, 2016. [55] X. Wu, R. Jiang, M.Q. Zhang, S. Li, Network-based global inference of human disease genes, Mol. Syst. Biol. 4 (1) (2008). [56] Z.-H. You, Z.-A. Huang, Z. Zhu, G.-Y. Yan, Z.-W. Li, Z. Wen, X. Chen, PBMDA: A novel and effective path-based computational model for miRNA-disease association prediction, PLoS Comput. Biol. 13 (3) (2017), e1005455. [57] J. Peng, W. Hui, Q. Li, B. Chen, J. Hao, Q. Jiang, X. Shang, Z. Wei, A learning-based framework for miRNA-disease association identification using neural networks, Bioinformatics (2019). [58] Y. Ding, F. Wang, X. Lei, B. Liao, F.-X. Wu, Deep belief network–Based Matrix Factorization Model for MicroRNA-Disease Associations Prediction, Evolutionary Bioinformatics 16 (2020) 1176934320919707. [59] J. Li, S. Zhang, T. Liu, C. Ning, Z. Zhang, W. Zhou, Neural Inductive Matrix Completion with Graph Convolutional Networks for miRNA-disease association prediction, Bioinformatics (2020). [60] X. Chen, D. Xie, L. Wang, Q. Zhao, Z.-H. You, H. Liu, BNPMDA: bipartite network projection for MiRNA–disease association prediction, Bioinformatics 34 (18) (2018) 3178–3186. [61] Y. Zhao, X. Chen, J. Yin, Adaptive boosting-based computational model for predicting potential miRNA-disease associations, Bioinformatics 35 (22) (2019) 4730–4738. [62] X. Chen, C.-C. Zhu, J. Yin, Ensemble of decision tree reveals potential miRNA- disease associations, PLoS Comput. Biol. 15 (7) (2019), e1007209. [63] X. Chen, J. Yin, J. Qu, L. Huang, MDHGI: Matrix Decomposition and Heterogeneous Graph Inference for miRNA-disease association prediction, PLoS Comput. Biol. 14 (8) (2018), e1006418. [64] H. Yu, X. Chen, L. Lu, Large-scale prediction of microRNA-disease associations by combinatorial prioritization algorithm, Sci. Rep. 7 (2017) 43792. [65] Z. Yang, F. Ren, C. Liu, S. He, G. Sun, Q. Gao, L. Yao, Y. Zhang, R. Miao, Y. Cao, dbDEMC: a database of differentially expressed miRNAs in human cancers, BMC Genom. (2010) S5. [66] B. Xie, Q. Ding, H. Han, D. Wu, miRCancer: a microRNA–cancer association database constructed by text mining on literature, Bioinformatics 29 (5) (2013) 638–644.