27 KiB
| title | date | updated | tags | categories | keywords | description | top_img | comments | cover | toc | toc_number | toc_style_simple | copyright | copyright_author | copyright_author_href | copyright_url | copyright_info | katex | highlight_shrink | aside |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| deepDR_ a network-based deep learning approach to in silico drug repositioning | 2021-03-05 08:48:36 | 2021-09-12 19:30:41 | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | true | <nil> | <nil> |
DeepDR:一种基于网络的深度学习方法在计算药物重新定位中的应用
摘要
动机:传统药物的发现和开发往往既耗时又高风险。 重新调整已批准药物的用途/重新定位为快速开发有效的治疗方法提供了一种相对低成本和高效率的方法。大规模、异质生物网络的出现为电子药物定位方法的发展提供了前所未有的机遇。然而,用大多数现有的药物定位方法捕捉高度非线性、异质的网络结构一直是具有挑战性的。 结果:在这项研究中,我们通过整合10个网络:1个药物疾病网络、1个药物副作用网络、1个药物靶点网络和7个药物药物网络,开发了一种基于网络的深度学习方法,称为深度DR,用于电子药物再利用。具体地说,DeepDR通过多模式深度自动编码器从异构网络中学习药物的高层特征。然后,学习到的药物的低维表示与临床报告的药物-疾病对一起通过可变自动编码器被集体编码和解码,以推断最初未被批准的已批准药物的候选药物。我们发现DeepDR显示出高性能[接收器操作特征曲线下的面积(AUROC) = 0.908],优于传统的基于网络或基于机器学习的方法。重要的是,深度DR预测的药物与疾病之间的关联得到了ClinicalTrials.gov数据库(AUROC.0.826)的验证,我们展示了几种用于阿尔茨海默病(例如利培酮和阿立哌唑)和帕金森病(例如哌甲酯和培高利特)的新型深度DR预测的批准药物。 可用性和实现:源代码和数据可从https://github.com/ChengF-Lab/deepDR 下载。 联系方式:chengf@ccf.org 补充信息:补充数据可在生物信息学网站上获得。
1 简介
长期以来,医学研究人员一直试图发现导致人类疾病的单分子缺陷,目标是开发“MagicBullet”靶向疗法。然而,这种“一基因、一种药物、一种疾病”的还原论信息范式忽视了疾病的内在复杂性,并继续挑战个性化诊断和药物发现(Greene和Loscalzo,2017)。最近的一项研究估计,制药公司在2015年花费了26亿美元来开发一种美国食品和药物管理局(FDA)批准的新药,而2003年这一数字为8.02亿美元(Avorn,2015)。虽然有许多因素导致这一有限的批准率,但一个重要的、经常被忽视的决定因素是继续坚持可追溯到Ehrlich工作的药物开发的经典“一基因、一种药物、一种疾病”假说(Tan和Grimes,2010)。由于药物靶点并不是孤立于组成其相关细胞分子结构的复杂蛋白质系统中发挥作用的,我们认为,每个药物-靶点相互作用都必须在适当的综合背景下进行研究(Cheng等人,2019年;Greene和Loscalzo,2017年)。这样做将对药物机制、潜在的不良影响和所谓的非目标效应提供新的见解,这些效应可用于合理地重新调整药物的用途,称为药物重新用途/重新定位(Cheng等人,2018年;Pushpakom等人,2018年)。然而,由于未知的复杂药理学和生物学基础,药物再利用充满了挑战。
在这些进步中,基于计算机科学的有效和智能算法通过整合大规模基因组和表型数据以及数百种已批准药物的化学和生物活性数据,为最初未获批准的多种复杂疾病的药物重新定位提供了公正、合理的路线图(Cheng等人,2018年;Pushpakom等人,2018年)。例如,不同的数据源和各种药物或疾病相似网络为预测新的药物-疾病关联提供了不同的信息和多层次的视角。因此,合并多个数据源可能会提高电子药物重新定位的准确性(Ching等人,2018年)。然而,大多数现有的药物重新定位方法仅限于药物相似网络、疾病相似网络或二元药物-疾病模型(Cheng等人,2012,2017)。此外,这些方法不能直接扩展以考虑不同生物网络之间的异质节点或网络拓扑信息(Cheng等人,2018年;Pushpakom等人,2018年)。
基于网络的信息特征在药物与疾病关系的预测中起着至关重要的作用。然而,在从毒品相关网络的异构数据源中保留网络结构的同时,学习信息丰富的低维网络表示(也称为网络嵌入)存在一些挑战。特别地,在这些网络中具有相同或相似功能注释的药物通常表现出复杂的混合关系,基于同源性(在网络中彼此非常接近)和结构相似性(相似的结构角色,而不管在网络中的位置)。因此,研究药物(节点)的低维嵌入在保持非线性网络结构的同时保持对新药物适应症的预测性是一项具有挑战性的任务。更具挑战性的是构建这样一种紧凑的低维药物嵌入,它在不同的药物功能和分子相互作用模式中是一致的,例如在不同类型的药物相关网络中是一致的(Gligorijevic等人,2018年)。
深度学习是一种很有前途的捕获复杂和高度非线性网络结构的技术,为许多场景提供了强大的工具,如语音识别、图像分类和自然语言处理,以及医学和生物学(Angermueller等人,2016年;Ching等人,2018年;Topol,2019年)。在这项研究中,我们开发了一种新的方法,称为深度学习基础上的药物重新定位(DeepdDR),系统地推断新的药物与疾病的关系,用于电子药物的再利用。DeepDR的基本概念是融合来自不同类型网络的不同信息,并推断现有药物的新应用,这些药物最初没有得到集体变异自动编码器(CVAE)的批准。深度DR的优点可以概括为:(I)深度DR集成了来自9个异构网络的各种信息,这可能会提高药物疾病预测的准确性,并为药物重新定位提供新的见解;(Ii)深度DR通过应用多层非线性函数来保留非线性网络结构,能够捕获多种类型网络的复杂拓扑模式;(Iii)深度DR使用边信息(药物特征)来预训练变化式自动编码器,通过同时提供评级和边来克服药物疾病评级稀疏性。我们发现,深度DR在从临床报道的药物-疾病网络中预测药物-疾病关联方面表现出很高的性能,超过了以前最先进的方法。重要的是,我们证明了DeepDR在从ClinicalTrials.gov数据库收集的外部验证集上有很高的准确性,这表明它具有很高的泛化能力。
2 材料和方法
2.1 异构网络重建
我们通过收集来自两个常用数据库的数据来收集临床报告或实验验证的药物疾病网络:DrugBank(Wishart等人,2018年)和repoDB(Brown和Patel,2017)。每种药物的化学名称、通用名称或商业名称通过医学主题词(MESH)和统一医学语言系统(UMLS)词汇表(Bodenreider,2004)进行标准化,并进一步从DrugBank数据库(v4.3)转换为DrugBank ID(Law等人,2014年)。每种疾病的通用名称由MESH注释。我们总共构建了9个网络:(I)临床报道的药物-药物相互作用,(Ii)药物-靶相互作用,(Iii)药物副作用关联,(Iv)化学相似性,(V)来自解剖治疗化学分类系统的治疗相似性,(Vi)药物靶序列相似性,(Vii)基因本体论(GO)生物学过程,(Viii)GO细胞成分和(Ix)GO分子功能。在补充材料中提供了关于建立不同种类的药物网络的更多细节。此外,还进一步收集了6677个临床报告的药物-疾病对,这些药物-疾病对连接了1519种药物和1229种疾病,用于构建预测性深度学习模型(补充表S1和S2)。对于外部验证集,我们从ClinicalTrials.gov数据库(https://clinicaltrials.gov/),)收集了最新的药物-疾病关联,方法是排除上述DrugBank(Wishart等人,2018年)和repoDB(Brown和Patel,2017年)数据库中的现有对。
2.2 基于随机游走的网络表示
我们采用曹(2016)中使用的方法来捕获网络结构信息,并表征每种药物的拓扑上下文。网络的顶点首先是随机排序的。
假设当前顶点是第i个顶点,转移矩阵A捕获不同顶点之间的转移概率。
它同时考虑了网络中的局部和全局拓扑连通性模式,以充分利用节点之间潜在的直接或间接关系。因此,在每个时刻,随机游走过程将以概率x继续,并将返回到原始顶点,并以概率1<EFBFBD>x重新开始该过程。该步骤导致递归关系如下:
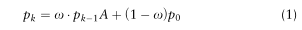 其中Pk是行向量,其第j个条目指示在k个转换步骤之后到达第j个顶点的概率,并且P0是初始的1热向量,第i个条目的值为1,而所有其他条目的值为0。通过对PK的每个随机游动求和,并对网络中的每个节点重复这一过程,我们可以得到一个概率共生矩阵。
其中Pk是行向量,其第j个条目指示在k个转换步骤之后到达第j个顶点的概率,并且P0是初始的1热向量,第i个条目的值为1,而所有其他条目的值为0。通过对PK的每个随机游动求和,并对网络中的每个节点重复这一过程,我们可以得到一个概率共生矩阵。
接下来,我们计算了一个移位的正点互信息(PPMI)矩阵(Bullinaria and Levy,2007)。PPMI矩阵可以被视为一种矩阵分解方法,其对共生矩阵进行分解以产生网络表示。PPMI矩阵可以按如下方式构建:
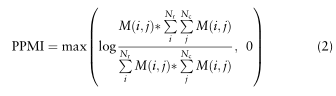 其中M是原始共生矩阵,Nr是行数,Nc是列数。负的PPMI值更改为0。基于随机游走的表示减轻了某些单独网络类型的稀疏性,该稀疏性在后续步骤中描述的更深层次的集成之前充当预处理步骤。
其中M是原始共生矩阵,Nr是行数,Nc是列数。负的PPMI值更改为0。基于随机游走的表示减轻了某些单独网络类型的稀疏性,该稀疏性在后续步骤中描述的更深层次的集成之前充当预处理步骤。
2.3多模深度自动编码器(MDA)网络融合
为了获得融合多个网络的高质量药物特征,我们遵循了之前提出的策略(Gligorijevic等人,2018年),该策略使用MDA集成了由PPMI矩阵表示的多个网络。通过使用多个非线性激活函数将n个药物的PPM I矩阵Xüj?2Rn<EFBFBD>n投影到共同的特征空间HC 2RDC<EFBFBD>n,MDA构造了n个药物的低维特征表示,该低维特征表示最佳地逼近所有网络。自动编码器是一种特殊类型的神经网络,由编码部分和解码部分组成(Vincent等人,2010年)。我们在以下几节中阐述了MDA的编码和解码部分。
2.3.1编码器
在模型的第一隐层中,我们首先计算了每个网络j~2F_1的低维非线性嵌入H_(?j?)encode 2Rdj_<EFBFBD>_n。。。;;Ng:
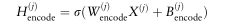 其中,W j??编码2Rdj?<EFBFBD>n和B?j?编码2?Rdj?<EFBFBD>n是加权矩阵和偏置矩阵。R是S形激活函数。
然后,我们将所有网络的非线性嵌入连接起来,并通过对它们应用多个非线性函数来计算公共特征表示。在得到公共表示之后,可以有L层。
其中,W j??编码2Rdj?<EFBFBD>n和B?j?编码2?Rdj?<EFBFBD>n是加权矩阵和偏置矩阵。R是S形激活函数。
然后,我们将所有网络的非线性嵌入连接起来,并通过对它们应用多个非线性函数来计算公共特征表示。在得到公共表示之后,可以有L层。
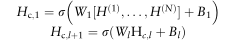 式中1/H 1?;。。。;H_N(?)<EFBFBD>是前几层的N个嵌入的级联激活矩阵,l_<EFBFBD>_(F1);。。;lg是连续集成嵌入的层号。
式中1/H 1?;。。。;H_N(?)<EFBFBD>是前几层的N个嵌入的级联激活矩阵,l_<EFBFBD>_(F1);。。;lg是连续集成嵌入的层号。
2.3.2解码器
2.4集体变分自动编码器
2.4.1 生成网络
虽然药物-疾病关联和药物特征是两种不同类型的信息,但是cVAE假设生成网络的输出根据输入的类型遵循不同的分布。我们将药物-疾病关联定义为Y,药物特征定义为X。按照VAE的一般做法,我们首先假设潜变量u和z遵循高斯分布:
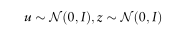 我在哪里<EFBFBD>Rk公司<EFBFBD>k是单位矩阵,k是潜在药物表征的维数。在积极的未标记(PU)学习框架的激励下,观察到的和未观察到的条目在目标中受到不同的惩罚(Elkan和Noto,2008;Hsieh等人,2014),我们引入了一个参数a来平衡正样本和负样本。当X和Y被输入同一个网络时,我们想通过不同的分布来区分它们。对于药物-疾病关联,所有药物的疾病j评级遵循伯努利分布:
我在哪里<EFBFBD>Rk公司<EFBFBD>k是单位矩阵,k是潜在药物表征的维数。在积极的未标记(PU)学习框架的激励下,观察到的和未观察到的条目在目标中受到不同的惩罚(Elkan和Noto,2008;Hsieh等人,2014),我们引入了一个参数a来平衡正样本和负样本。当X和Y被输入同一个网络时,我们想通过不同的分布来区分它们。对于药物-疾病关联,所有药物的疾病j评级遵循伯努利分布:
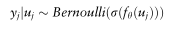 这定义了将药物-疾病关联作为输入时的损失函数,即疾病j的逻辑对数似然率:
这定义了将药物-疾病关联作为输入时的损失函数,即疾病j的逻辑对数似然率:
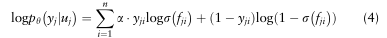 其中,Fji是向量fh Iuj?的第i个元素,Fh Ig uj?通过Sigmoid函数进行归一化,使得Fji在(0,1)内。
其中,Fji是向量fh Iuj?的第i个元素,Fh Ig uj?通过Sigmoid函数进行归一化,使得Fji在(0,1)内。
2.4.2 推断网络
3结果
3.1 deepDR管道
如图1所示,深度DR由三个步骤组成:(I)深度DR首先应用随机游走与重启(RWR)方法,然后构建捕获网络结构信息的PPMI矩阵,从而将每个网络的拓扑结构转换为高质量的矢量表示。(Ii)深度DR以无监督的方式将每个网络的PPMI矩阵融合成对所有使用MDA的网络通用的紧凑的低维特征表示。在Depth自动编码器的早期部分,DeepDR使用单独的层来处理每种网络类型;之后它将所有层连接到单个瓶颈层。因此,我们可以从这个单一的瓶颈层中提取高质量的特征。(Iii)深度DR使用CVAE推断药物和疾病之间的潜在联系。具体地说,它将第二步提取的高质量特征馈送到VAE进行预训练,然后通过馈送药物-疾病关联网络来细化VAE。
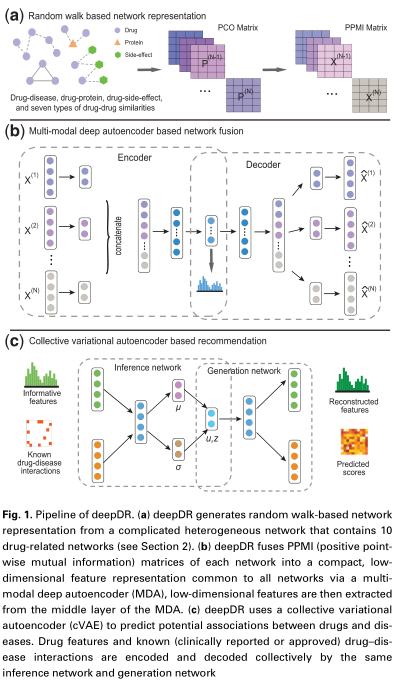 图1所示。deepDR的管道。(a) deepDR从一个包含10个药物相关网络的复杂异构网络中生成基于随机漫步的网络表示(见第2节)。通过多模态深度自编码器(MDA)来表示所有网络共同的低维特征,然后从MDA的中间层提取低维特征。(c) deepDR使用集体变分自编码器(cVAE)来预测药物和疾病之间的潜在关联。药物特征和已知(临床报告或批准)药物-疾病相互作用通过同一推理网络和生成网络共同编码和解码
图1所示。deepDR的管道。(a) deepDR从一个包含10个药物相关网络的复杂异构网络中生成基于随机漫步的网络表示(见第2节)。通过多模态深度自编码器(MDA)来表示所有网络共同的低维特征,然后从MDA的中间层提取低维特征。(c) deepDR使用集体变分自编码器(cVAE)来预测药物和疾病之间的潜在关联。药物特征和已知(临床报告或批准)药物-疾病相互作用通过同一推理网络和生成网络共同编码和解码
3.2 Baseline methods
预测结果是基于下面列出的模型之间的详细比较。 更多的基线方法和超参数选择的细节(补充表S6-S9)可以在补充材料中找到。 •DTINet: DTINet (Luo et al., 2017)侧重于从异构网络中学习特征的低维向量表示,然后应用归纳矩阵补全(IMC) (Nagarajan and Dhillon, 2014)基于学习到的表示进行预测。 KBMF:核贝叶斯矩阵分解方法(Go¨nen et al., 2013)可以利用多个侧信息源,可以应用于推荐系统。 支持向量机(SVM):支持向量机(Cortes and Vapnik, 1995)基于统计学习理论,从结构风险最小化原理和Vapnik - chervonenkis (VC)维度推导而来。 RWR:随机漫步与重启(Cao等,2014;Ko¨hler et al., 2008),这是一种网络扩散算法,用于测量网络的两个节点之间的接近程度。 Katz: Katz (Singh-Blom et al., 2013)度量是一种基于图的方法,通过计算节点之间存在多少不同的路径长度来寻找节点与给定节点的相似性。
3.3 deepDR在交叉验证上的性能
为了评估DeepDR的性能,我们首先进行了5次交叉验证。我们总共收集了6677个临床报道的药物-疾病对,连接了1519种批准的药物和1229个人类疾病术语。在5次交叉验证中,我们随机选择了20%的临床报告药物-疾病对的子集和匹配数量的随机抽样的未知对作为测试集,其余80%的临床报告的未知对被随机抽样的相同数量的药物-疾病对用于训练模型。使用接收器工作特征曲线下面积(AUROC)和精确召回曲线下面积(AUPR)来评价DeepDR的整体性能。为了减少交叉验证的数据偏差,重复10次,并计算平均性能。我们发现DeepDR在5次交叉验证中表现出很高的准确性(AUROC?0.908和AURC?0.923),优于最先进的方法:DTINET(AUROC?0.862和AUROC?0.892)、KBMF(AUROC?0.791和AUROC?0.826)、RF(AUROC?0.783和AUROC?0.805)、支持向量机(AUROC?0.771和AUROC?0.778)。RWR(AUROC?0.708和AUROC?0.734)和CATZ(AUROC?0.724和AUPR?0.741)(图2a和b,补充表S5)。
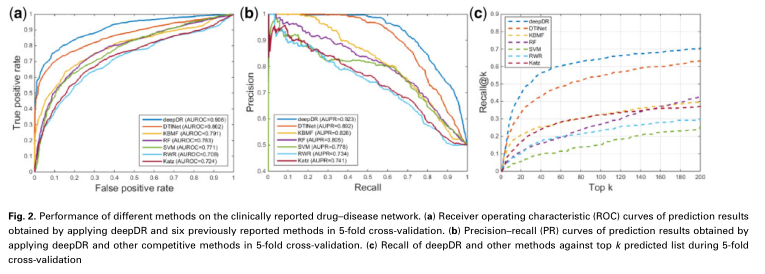 由于正确预测的真阳性的数量反映了预测方法区分真阳性的辨别能力,特别是当阴性样本的数量远远大于阳性样本的数量时,我们进一步使用‘recall@top-k’作为评估度量,其定义为在药物的top-k预测列表中检索到的真实批准的疾病(适应症)的比例。使用该度量的动机是,通常需要一种能够准确地恢复top-k预测列表中的真实相互作用疾病的方法,并且该方法对于下游实验验证是有用的。如图2c所示,对于前200名预测候选,深度DR的召回率为70%,显著优于DTINet(62%)、KBMF(40%)、RF(43%)、SVM(25%)、RWR(30%)和Katz(38%)。
由于正确预测的真阳性的数量反映了预测方法区分真阳性的辨别能力,特别是当阴性样本的数量远远大于阳性样本的数量时,我们进一步使用‘recall@top-k’作为评估度量,其定义为在药物的top-k预测列表中检索到的真实批准的疾病(适应症)的比例。使用该度量的动机是,通常需要一种能够准确地恢复top-k预测列表中的真实相互作用疾病的方法,并且该方法对于下游实验验证是有用的。如图2c所示,对于前200名预测候选,深度DR的召回率为70%,显著优于DTINet(62%)、KBMF(40%)、RF(43%)、SVM(25%)、RWR(30%)和Katz(38%)。
3.4 DeepDR在外部验证集上的性能
对回顾性数据进行交叉验证可能会导致过于乐观的结果。为了进行客观的绩效评估,我们进一步从ClinicalTrials.gov数据库中收集了临床报道的药物-疾病对,作为外部验证集。外部验证集包含129个以前未使用过的新发现的关联。图3a给出了此任务的所有方法的性能比较。深度DR的性能优于其他方法。例如,DeepDR的AUROC值为0.826,明显优于DTINet(0.732)、KBMF(0.673)、RF(0.788)、支持向量机(0.753)、RWR(0.601)和CATZ(0.563)。此外,我们还显示了与给定的顶级阈值相关的正确预测的药物-疾病关联的召回率,如图3b所示。具体地说,DeepDR(回忆率为25%)在几乎每个最高排名200的阈值上都比其他方法获得了更准确的药物-疾病关联预测,与5倍交叉验证一致,表明它具有很高的泛化能力。
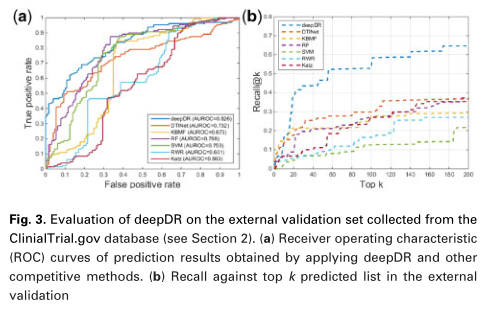 图3.对从ClinialTrial.gov数据库收集的外部验证集进行的深度DR评估(请参见第2节)。(A)应用深度DR和其他竞争性方法得到的预测结果的接收器工作特性(ROC)曲线。(B)根据外部验证中的前k个预测列表重新调用。
图3.对从ClinialTrial.gov数据库收集的外部验证集进行的深度DR评估(请参见第2节)。(A)应用深度DR和其他竞争性方法得到的预测结果的接收器工作特性(ROC)曲线。(B)根据外部验证中的前k个预测列表重新调用。
3.5烧蚀分析的DeepDR性能
深度DR主要由MDA和CVAE两部分组成。为了检验每种成分的贡献,我们将深度DR与几种组合进行了比较。首先,我们将MDA与另外两种网络表示方法DeepWalk(Perozzi等人,2014)和SDNE(Wang等人,2016)进行了比较,以检查MDA的贡献。DeepWalk通过截断随机行走将图转换为线性序列的集合,并利用跳跃语法模型来学习顶点的低维表示(Perozzi等人,2014年)。SDNE是一种深度网络表示模型,它使用深度自动编码器考虑一阶和二阶邻近度(Wang等人,2016年)。如图4a和b所示,我们发现MDA优于DeepWalk和SDNE(图4a和b)。具体地说,MDA和SDNE都是基于深度学习的嵌入方法,它们的性能都优于DeepWalk。如前所述,SDNE不能考虑不同网络之间的内部交互。在本研究中,MDA能够将不同的信息融合成所有网络通用的低维特征表示,并在本实验中取得最佳性能。为了进一步检验CVAE的贡献,我们使用从MDA或传统的主成分分析(PCA)中提取的相同特征,将深度DR与SVM和RF进行了比较。具体地说,我们比较了深度DR与四种不同的组合,包括MDA塔RF,MDA塔支持向量机,PCA塔RF,PCA塔支持向量机,这些组合的评估结果如图5a和b所示,我们发现深度DR取得了最好的性能。由于缺乏与疾病相关的信息,传统的分类器表现不佳,而深度DR充分利用了多种与药物相关的信息,不需要与疾病相关的网络,这就是它优于其他方法的原因。

3.6深部DR的药理学解释
基于深度神经网络的网络嵌入能够将与预测任务相关的复杂特征关系编码到顶点的向量表示中。评估顶点表示质量的一种直观方式是通过可视化,嵌入在学习特征中的大部分知识由特征矩阵编码。通过可视化特征矩阵,我们可以解开复杂的网络。我们使用t-SNE(t分布随机邻居嵌入)(van der Maten和Hinton,2008)研究了通过网络嵌入学习到的内部特征,t-SNE是一种非线性降维方法,它将高维空间中的相似点嵌入为二维中的近似点。图中的每个点表示从深度DR框架中的MDA中间层提取的900维特征向量投影的药物节点。使用相同的颜色高亮显示相同类型的节点。在相同设置下,不同颜色组之间边界清晰的群集表示更好的表示。通过t-SNE,我们将按解剖治疗化学(ATC)分类系统代码的第一级分组的药物投影到2D空间。我们发现,MDA生成的特征向量能够很好地区分按ATC代码分组的14种药物(附图2)。S1a),明显优于PCA方法(补充图。S1b)。 总而言之,DeepdR显示了高度的网络嵌入能力,以保持异质药物-疾病网络的固有属性和结构。
3.7案例研究:通过计算确定阿尔茨海默病和帕金森病的批准药物
为了进一步验证深度DR的预测能力,我们对阿尔茨海默病(AD)和帕金森病(PD)这两类尚无有效治疗手段的神经退行性疾病进行了个案研究。 阿尔茨海默病(AD)。我们重点关注了补充表S3中AD的前20个深度DR预测候选对象。对于每种药物,我们都显示了标准名称、预测得分和文献报道的证据。 异丙肾上腺素是一种非选择性肾上腺素受体激动剂,被批准用于治疗心动过缓和心脏阻滞(Sato等人,2004年)。在此,异丙肾上腺素是潜在治疗AD的首选候选药物。先前的临床研究报告了异丙肾上腺素可以减少阿尔茨海默病患者的淀粉样斑块(Ohm等人,1991)。多巴胺是儿茶酚胺和苯乙胺家族的一种化合物,在人脑中起着重要作用,深度DR预测它与阿尔茨海默病(AD)有关。这一预测可以得到之前一项研究的支持,该研究表明大脑中缺乏多巴胺可能会导致阿尔茨海默氏症的一些早期症状(Li等人,2004年)。 利培酮是一种非典型的抗精神病药物,主要用于治疗精神分裂症和双相情感障碍,据Deepth DR预测,它对阿尔茨海默病也有潜在的疗效。这一预测得到了先前文献的支持(Katz等人,2007年;Negron和Reichman,2000年)。此外,DeepDR发现阿立哌唑--另一种主要用于治疗精神分裂症和双相情感障碍的非典型抗精神病药物--与阿尔茨海默病有关,这得到了多项证据的支持(De Deyn等人,2005年、2013年)。在按可信度排名的前20种预测药物中,有14种药物(成功率为70%)通过临床研究、临床前研究和其他文献数据的各种证据进行了验证(补充表S3)。 帕金森氏病(PD)。我们重点关注了补充表S4中关于PD的前20个深度DR预测候选者。我们发现,20种药物中有14种(成功率为70%)通过了先前的文献研究(补充表S4)。例如,深度DR预测,乙醇胺类抗胆碱能药物奥非那林与帕金森病有关联。这样的预测可以得到以前的文献的支持(Bassi等人,1986;Strang,1964)。培高利特是一种以麦角林为基础的多巴胺受体激动剂,与大脑黑质中多巴胺活性降低有关,深度DR预测它与帕金森病有关,这与之前的临床前和临床研究一致(Goetz等人,1983年;Storch等人,2005年;Van Camp等人,2004年)。此外,用于治疗注意力缺陷多动障碍(ADHD)和发作性睡病的一种兴奋剂药物哌醋甲酯,被预测与帕金森病有关,并得到了多项研究的支持(Auriel等人,2009年;Devos等人,2013年;Mendonc?a等人,2007年)。 总而言之,Deepre DR为优先考虑治疗阿尔茨海默病和帕金森病的潜在再利用药物提供了一个有用的工具。
4讨论和结论
在这项研究中,我们提出了一个新的深度学习框架DeepdR来揭示药物和疾病之间的潜在联系。除金标准药物-疾病关联网络外,我们还整合了1个药物-药物相互作用网络、1个药物-蛋白关联网络、1个药物副作用关联网络和6个药物-药物相似网络,构建了一个包含多种信息的复杂异构网络,并以多视角预测新的药物-疾病关联。DeepDR首先将来自众多不同网络类型的不同信息融合成紧凑的低维特征表示,然后将学习到的药物特征的低维表示与已知的药物-疾病相互作用对一起馈入变化式自动编码器,以预测新的药物-疾病关联。理论上,由于采用了多模式深度自动编码器(MDA)来捕捉不同数据源的复杂拓扑模式,DeepDR优于现有的药物定位方法。此外,深度DR能够通过应用多层非线性函数来保持非线性网络结构。 它还用药物特征补充稀疏评级,因为向同一VAE提供辅助信息增加了用于训练的样本数量,这也类似于训练前的步骤。具体地说,毒品与疾病的联系和毒品特征是不同的信息源,都是与毒品相关的信息,因此可以通过同一推理网络和生成网络对其进行集体编码和解码。我们从交叉验证、外部验证和案例研究三个方面验证了深度DR的预测能力,结果表明,我们的方法在发现新的药物-疾病关联方面取得了最先进的性能。深度DR充分利用了多个药物相关网络,而RWR和KATZ等其他方法都需要一个药物相似网络和一个疾病相似网络。为了与其他方法进行比较,我们从疾病-蛋白质关联网络的Jaccard相似度计算出疾病相似度网络,并为这些方法选择药物化学相似度,因此在比较过程中可能会有一定的偏差。在未来的研究中,由于DeepDR是一个可扩展的框架,从更多的数据库和文献中收集和整合更多相关的关联数据可能会提高其能力。此外,由于CVAE的结构,目前深度DR只能集成与药物相关的信息,未来的发展方向是修改神经网络推荐的结构,扩展深度DR以集成与药物相关的信息和与疾病相关的信息。 我们承认,在当前基于网络的深度学习框架下,深度DR存在一些潜在的局限性。尽管我们的大量工作从公开的数据库中收集了大规模的、实验报道的药物-靶标相互作用,但数据质量也不能得到保证,网络数据可能是不完整的。例如,由于公开可用的数据库和已发表的文献中缺乏阴性药物-疾病对,因此在机器学习研究中构建黄金标准的未知对作为阴性样本一直是具有挑战性的。我们在补充数据库S1中提供了深度耐药预测药物-疾病对的完整列表。未来有必要对患者数据(例如健康保险索赔数据)进行最先进的药物流行病学分析,并对深度DR预测的候选者进行体外或体内机制研究。 综上所述,我们的发现表明,在电子计算机中,药物再利用可能受益于基于网络的深入学习,探索药物-靶点-疾病异构网络之间的关系。从翻译的角度来看,如果广泛应用,这里开发的网络工具可以从基于网络的药物再利用的角度帮助开发针对多种复杂疾病的新颖、有效的治疗方法。