34 KiB
| title | date | updated | tags | categories | keywords | description | top_img | comments | cover | toc | toc_number | toc_style_simple | copyright | copyright_author | copyright_author_href | copyright_url | copyright_info | katex | highlight_shrink | aside |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MultiDTI_ drug–target interaction prediction based on multi-modal representation learning to bridge the gap between new chemical | 2021-12-29 14:29:57 | 2022-02-07 09:14:58 | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | true | <nil> | <nil> |
MultiDTI:基于多模态表示学习的药物-靶相互作用预测,以弥合新化学实体和已知异构网络之间的差距
Deshan Zhou 1, Zhijian Xu 2,, WenTao Li3, Xiaolan Xie4, and Shaoliang Peng1,3,* 1Department of Computer Science, Hunan University, Changsha 410082, China, 2CAS Key Laboratory of Receptor Research, Drug Discovery and Design Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China, 3Department of Computer Science, National University of Defense Technology, Changsha 410073, China and 4College of Information Science and Engineering, Guilin University of Technology, Guilin 541004, China *To whom correspondence should be addressed. Associate Editor: Teresa Przytycka Received on February 24, 2021; revised on May 27, 2021; editorial decision on June 21, 2021; accepted on June 27, 2021
Abstract
动机:预测新药与靶点的相互作用是新药开发、了解其副作用和药物重新定位的重要步骤。异构数据源可以为药物-靶点相互作用预测提供全面的信息和不同的视角。因此,有许多计算方法依赖于异构网络。它们大多使用图形相关算法来描述异构网络中的节点,以预测新药-靶点相互作用(DTI)。然而,这些方法只能在已知的异构网络数据集中进行预测,不能支持对异构网络之外的新化学实体的预测,这阻碍了药物的进一步发现和开发。 结果:为了解决这个问题,我们提出了一个名为“MultiDTI”的多模式DTI预测模型,该模型使用了我们提出的基于异构网络的联合学习框架。它将异构网络的交互或关联信息与药物/靶点序列信息相结合,将异构网络中的药物、靶点、副作用和疾病节点映射到一个公共空间。通过这种方式,“MultiDTI”可以根据新实体的化学结构,将新的化学实体映射到这个已知的公共空间。也就是说,弥合新化学实体和已知异构网络之间的差距。我们的模型具有很强的预测性能,在10倍交叉验证下,该模型的接收器工作特性曲线下的面积为0.961,精确召回曲线下的面积为0.947。此外,一些预测的新DTI已被ChEMBL数据库确认。我们的研究结果表明,“多重DTI”是预测新DTI的一个强大而实用的工具,它可以促进药物发现或药物重新定位的发展。
Availability and implementation: Python codes and dataset are available at https://github.com/Deshan-Zhou/ MultiDTI/. Contact: slpeng@hnu.edu.cn or zjxu@simm.ac.cn or xie_xiao_lan@foxmail.com. Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
药物-靶标相互作用 (DTI) 的鉴定引起了极大的关注,因为它是新药开发,了解其副作用和药物重新定位的重要步骤 (Ding等,2014)。大多数DTI是由与内源性小分子竞争蛋白质上功能所需结合位点的药物形成的 (Hopkins和Groom,2002),进而影响疾病状况。目前市场上有数千种FDA批准的药物,以及处于临床试验后期阶段的药物 (Xia等,2010)。这些药物可能具有潜在的未观察到的靶标和未知的适应症。然而,通过生物实验确定dti是费时、费力且昂贵的 (Zheng等,2020)。因此,建立一个计算模型的强烈动机,该模型可以估计新的药物-靶标对的相互作用,并为生物学家提供DTI候选物,以减少湿实验室实验的工作量。
传统的基于计算机的DTI预测方法主要包括基于对接的方法 (Garrett,2009) 和基于配体的方法 (Michael,2007)。基于对接的方法需要目标蛋白的三维结构。这些方法具有局限性,因为并非所有目标结构信息都是已知的。基于配体的方法使用相互作用配体的已知规则来进行预测。如果已知配体的数量不足,这些方法将无法很好地工作。
近年来,通过计算预测DTI的方法主要有基于相似度的方法和基于网络的方法。基于相似性的方法依赖于具有相似结构的化合物可能具有相似性质的假设。早期的方法使用基于一对药物-药物和基因-基因相似性度量的药物-基因关联评分方案,结合逻辑回归组件来整合多个指标的分数以产生最终的关联分数(Perlman et al. , 2011)。早期的基于相似性的方法大多使用手工制作的特征。近年来,研究表明,将特征学习能力集成到机器学习模型中可以提高预测性能(Abbasi et al., 2020)。最近,深度学习方法受到越来越多的关注,因为它们似乎克服了某些限制,减少了预测过程中特征信息的损失(Maryam et al., 2020)。 'DeepDTIs'(Wen et al., 2017)首先从简单的化学子结构和序列信息中自动提取药物和靶标的特征,然后使用有效的深度学习方法——深度信念网络(Deep Belief Network,DBN)构建分类模型。 “DeepSSDTIs”(Bahi 和 Batouche,2018 年)基于深度半监督学习,使用大规模化学蛋白质数据准确预测潜在的新 DTI。然而,这些方法的输入是药物和靶标的分子指纹或分子描述符,没有考虑原子之间的局部连接和氨基酸的局部化学结构。最先进的基于相似性的方法使用药物和蛋白质序列的一维表示,并使用强大的高级深度学习模型来提取复杂的局部化学信息和序列中局部结构之间的上下文关系(Abbasi et al., 2020; Chen et等人,2020;Huang 等人,2020;Karimi 等人,2019;Nguyen 等人,2020;Quan 等人,2019;Tsubaki 等人,2019)。最后将药物和靶标的特征信息拼接起来输入神经网络进行预测。上面列出的一些方法将药物的一维表示作为特征表示的图。其他基于相似相似性的方法(Lee et al., 2019; Torng and Altman, 2019; Zheng et al., 2020)虽然没有完全使用药物或靶点的一维序列表示,但本质上是第一个特征提取药物和靶点的表示。之后,基于具有相似结构的化合物应该具有相似性质的假设,将提取的药物和靶标特征拼接并输入神经网络进行DTI预测。一般来说,基于相似性的方法主要是打算通过药物靶点对的特征来预测药物靶点对是否相互作用。重点是寻找更先进、性能更好和更合理的特征提取器。这类方法的优点是基于生物学特性,可扩展性比较强,在模型泛化能力强的情况下,可以预测数据集外的药物靶点相互作用。但是,这种方法也存在一些问题。它只考虑药物靶点对的生物结构信息来预测它们的相互作用,考虑的信息相对单边。此外,一些模型(Abbasi et al., 2020; Chen et al., 2020; Nguyen et al., 2020; Quan et al., 2019)显示固定蛋白长度设置为1000左右,蛋白序列Drugbank 数据库中的长度可以长达 14 507。这些模型无法预测数据集之外的长序列药物靶标对(图 1)。
基于网络的方法首先构建一个包含药物和目标的网络,并使用基于图的技术根据已知边预测未知边。 主要思想是药物倾向于与相似的靶标结合,反之亦然(Luo et al.,2017)。 早期的方法使用基于网络的推理 (NBI) 方法,该方法仅使用药物-靶点二分网络拓扑相似性来推断已知药物的新靶点 (Cheng et al., 2012)。 埃扎特等人。 (2016) 使用图正则化的矩阵分解方法以二分图作为输入来预测 DTI。 最近,随着与药物/靶点相关的数据源越来越多,异构数据源可以为预测药物-靶点相关性提供更多信息和不同视角。 研究表明,药物和靶点的特性也可以通过它们在生物系统中的各种功能作用来表征(例如,蛋白质-蛋白质相互作用和药物-疾病关联)(Wan et al., 2019)。 整合来自异构数据源的各种信息可以进一步提高 DTI 预测的准确性(Luo et al.,2017)。 刘等人。 应用带重启的随机游走 (RWR) 来预测药物靶向异构网络上的 DTI。 曾等人。 使用多模式深度自动编码器从异构网络中学习药物的高级特征来预测 DTI。 在过去的几年里,图神经网络的概念、操作和模型一直在不断发展和发展(Wu et al., 2020)。 并且图卷积神经网络在提取嵌入式图形方面表现出了很强的性能(Schlichtkrull et al.,2017; Sun et al., 2020a,b; Zitnik et al., 2018),极大地促进了药物-靶点相互作用预测的发展 . 赵等人。 (2021)和孙等人。 (2020a,b) 使用图卷积神经网络来学习药物并针对异构网络嵌入。 但这类方法主要是对固有异构网络的节点进行表征,对于异构网络之外的药物,很难实现目标预测。 此外,节点的特征表示主要基于异构网络中的拓扑信息,没有深入考虑药物和靶点的生物结构信息。
在本文中,为了解决基于相似性和基于网络的方法的问题,我们提出了一种新的端到端方法,称为“MultiDTI”,用于基于多模态表示从异构网络中预测新的 DTI。 在表示学习领域,“模态”一词是指特定的信息编码方法或机制,涉及多模态的表示学习任务将具有多模态特征(Guo et al., 2019)。 “MultiDTI”使用我们提出的基于异构网络的联合表示框架将基于相似性的方法与基于网络的方法相结合。 首先,它以药物和靶标的一维序列信息为输入,通过低复杂度的词级深度卷积神经网络自动学习具有较长固定输入长度的序列特征(Johnson and Zhang,2017)。 然后,我们提出了一种基于异构网络的联合表示框架,将异构网络中的药物、靶点、副作用和疾病的表示投影到一个公共空间中。 最后,它根据公共空间中药物与目标之间的距离预测新的 DTI。 将“MultiDTI”与基于相似性的方法进行比较,不仅深入挖掘了药物和靶点的结构信息,还考虑了药物、靶点、副作用和疾病等异构网络中的相互作用。 此外,该模型利用5.1.7版本的Drugbank数据库中的所有药物和靶序列信息构建序列片段词汇表,并将模型的固定序列长度设置为所有序列中最长的长度,使得 模型可以预测长序列药物-靶点对。 将“MultiDTI”与基于网络的方法进行比较,它深入挖掘了药物和靶序列的复杂结构信息,可以预测异构网络数据集之外的药物潜在靶点。 该模型的数据集基于异构网络,异构网络中所有药物和靶点的序列信息均从 Drugbank 数据库中下载。 我们的端到端预测模型的预测性能明显优于使用相同异构网络数据集的其他模型。 此外,“MultiDTI”模型预测的一些新的 DTI 已经被最新版本的 ChEMBL 数据库验证,进一步证明了我们的模型具有很强的预测能力。 上述结果表明,‘MultiDTI’是预测潜在 DTI 的强大而实用的工具。 它可以为生物学家提供DTI候选者,以减轻湿实验室实验的工作量,促进新药开发和药物重新定位的发展。
总之,本文的主要贡献如下:
- 我们提出了第一个端到端的深度学习模型,它结合了两种主流的基于相似性的方法和基于网络的方法来预测潜在的 DTI。
- 我们提出了基于异构网络的联合表示框架,以充分利用药物/靶点的序列结构信息以及包含药物、靶点、副作用和疾病的异构网络中的相互作用或关联信息。
- 模型支持长序列DTI预测,可以预测数据集外的药物潜在靶点,提高了模型的适用性。
- 我们的模型在评价指标上优于一些最先进的药物-靶点相互作用预测方法,部分预测结果被验证是正确的。
2 Materials and methods
2.1 Datasets
2.1.1 Heterogeneous network
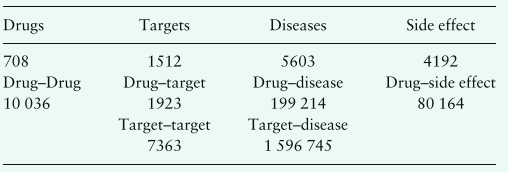
我们使用之前论文(Wan et al., 2019)的部分异构网络数据集,其中包含六个相关网络:药物-蛋白质相互作用网络、药物-药物相互作用网络、蛋白质-蛋白质相互作用网络、药物-疾病关联网络、 蛋白质-疾病关联网络和药物副作用关联网络。 其中,药物-靶点相互作用网络和药物-药物相互作用网络提取自 Drugbank 3.0 版(Knox et al., 2011)。 目标-目标交互网络是从 HPRD 数据库 Release 9 (Keshava Prasad et al., 2009) 中提取的。 药物-疾病关联网络和目标-疾病关联网络是从比较毒物基因组学数据库中提取的(Davis et al., 2013)。 药物副作用关联网络是从 SIDER 数据库第 2 版中提取的(Kuhn 等人,2010)。 这个异构网络包含 708 个药物节点、1512 个目标节点、5603 个疾病节点和 4192 个副作用节点。 它具有二元边权重,即 1 表示已知的交互或关联,0 表示未知或没有交互或关联。 更多细节可以在表 1 中找到。
2.1.2 Sequences of drug and target
异质网络中的药物由 SMILES 序列表示,异质网络中的靶点由原始蛋白质序列表示。 这些序列主要是通过 Drugbank ID 和 UniProt ID 从 Drugbank 数据库中获得的。 Drugbank 数据库中未找到的从 PubChem 数据库和 UniProt 数据库下载。
2.2 Drug and target representation
我们使用 n-gram 嵌入技术来定义蛋白质序列中的“单词”,也就是说,我们将这些蛋白质序列划分为重叠的 n-gram 氨基酸。给定一个蛋白质序列$T=s_{1}, s_{2}, \ldots, s_{T}$,其中si是第$i^{th}$个氨基酸,$T$是序列长度,target可以表示为:$T=\left(s_{1}, \ldots, s_{n}\right),\left(s_{2}, \ldots, s_{n+1}\right), \ldots,\left(s_{T-n+1}, \ldots, s_{T}\right)$。这里的$\left(s_{i}, \ldots, s_{i+n-1}\right)$是代表蛋白质序列的第i个单词。因为总共有20个氨基酸,所以单词总数是$20^{n}$。为了将词汇量控制在合适的范围内,避免出现低频词,我们将n设置为3。
为了增加模型的适用性,我们在构建蛋白质词汇表时,不仅考虑了异构网络数据集中的蛋白质序列,还考虑了 Drugbank 数据库中的所有蛋白质序列。 因此,蛋白质序列的最大固定长度$L_{max}$被认为是15000,较短长度的蛋白质用零填充。 最后,蛋白质序列的嵌入表示是$T_e \in R^{L_{max} \times E_v}$,其中 $E_v$表示单词在蛋白质词汇表中的嵌入向量。
药物分子由包含一系列字符的 SMILES 字符串表示。 与蛋白质序列处理不同,$D=a_{1}, a_{2}, \ldots, a_{D}$ 用于表示药物,其中$a_i$表示原子或结构指示符,$D$是药物序列的长度。 根据 Drugbank 数据库中最长的药物序列长度,我们将药物序列的最大固定长度 $l_{max}$设置为 1500,长度较短的药物用 0 填充。 最后,药物序列的嵌入表示为$D_e \in R^{l_{max} \times e_v}$,其中 $e_v$表示原子或结构指示符的嵌入向量。
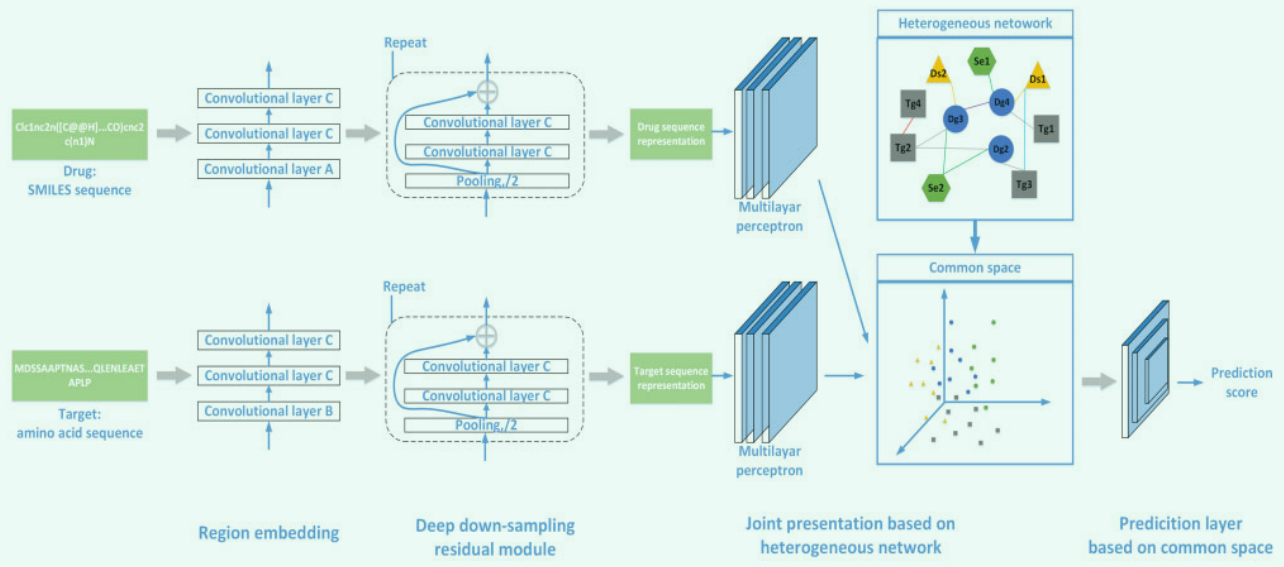
2.3 Deep convolutional neural network for long sequences
在获得药物和目标的长序列的原始嵌入表示后,我们使用计算复杂度较低的深度金字塔 CNN 模块 (Johnson and Zhang, 2017) 有效地表示长序列中的长距离关联和更全局信息。
2.3.1 Sequence region embedding
当我们表征一个长序列时,我们首先需要表征序列片段。 总之,让每个word embedding不仅包含自己的信息,还包含左右邻居的信息。 我们使用卷积层来学习这个区域嵌入。 这个卷积层由多个卷积核组成,每个卷积核响应每个输入的一个小区域。 给定一个序列$S \in R^{l \times e_v}$的原始嵌入表示,第n个卷积核和第i个区域的计算表达式如下: $S_{i, n}^{\prime}=W_{n} \odot S_{i, 1}+b_{n}$ 其中 $S_{i,1} \in R^{3\times e_v}$ 表示原始嵌入表示 S 在第 1 个通道中的第 i 个区域表示。这里,我们将卷积核的数量固定为 N,并且内核在序列方向上的大小( 覆盖的小区域)固定为3。因此,这里的第i个区域是一个范围为3的区域,由第i个单词与左右邻居组成。 $W_n \in R^{3 \times e_v}$和 $b_n \in R_1$分别代表第n个卷积核的权重和偏差。 然后经过n个卷积核的卷积层处理后,序列的表示就变成了$S^{\prime}\in R^{(l-2) \times n}$。
接下来,我们使用具有相同输入和输出通道数的两层卷积层来允许区域嵌入与其邻居交互。 与原来的深度金字塔 CNN 模块不同,我们这里没有使用快捷连接模块,更注重区域的嵌入。给定首尾加零序列的嵌入表示$S^{\prime} \in R^{l \times n}$,第m个卷积核和第i个区域的计算表达式如下: $S_{i, m}^{\prime \prime}=\sum_{i=1}^{n}\left(W_{m} \odot \operatorname{ReL} U\left(S_{i, n}^{\prime}\right)+b_{m}\right)$ 公式3 其中 $S^{\prime}_{i,n} \in R^{3 \times 1}$表示 $S^{\prime}$ 的第 n 个通道中的第 i 个区域表示。$W_m \in R^{3 \times 1}$和 $b_n \in R^1$分别代表第m个卷积核的权重和偏置。 $ReLU(\cdot)$表示非线性激活函数,这里使用预激活。 在处理两个相同的非线性卷积层后,序列表示为$S^{\prime\prime\prime} \in R^{l-2) \times n}$。
2.3.2 深度下采样残差模块
在这一步中,我们使用固定特征映射数量的降采样,以及预激活的快捷连接,以提供与长序列的长期关联,从而获得更多的全局信息。深度下采样残差模块由多个下采样残差层组成,每个下采样残差层将序列在长度方向上的内部表示减少了一半。经过多次降采样残差层处理后,序列在长度方向上的内部表示将变为1。. 给定上一层得到的序列表示$S^{(i)}$,下采样残差层处理的结果表示为: $S^{(i+1)}=\operatorname{down}\left(S^{(i)}\right)+f\left(\text { down }\left(S^{(i)}\right)\right)$ 其中$S^{(0)}=S^{\prime \prime \prime} . d \text { own }(\cdot)$表示大小为3,步长为2的最大池化函数。 也就是说,它通过将最大值嵌入到序列长度方向上跨度为2 的三个连续区域中来生成序列的新嵌入表示。 $f(\cdot)$表示公式3对应的两层卷积层。
函数$f(\cdot)$中先非线性激活再线性加权的方法,使得公式4包括加法的结果也是线性加权的结果。直观地说,这种“线性”简化了深度网络的训练。 另外,在特征图数量固定的情况下,每次序列embedding由一个下采样残差层处理,计算时间随着序列embedding大小减半而减半。深度下采样残差模块的总计算时间被限制在一个恒定的范围内——所花费的时间是$S^{(0)}$计算结果的两倍,这使得它非常适合长序列表征。 最后,我们完成了序列的特征提取,表示为$S^{fin} \in R^n$。
2.4 基于异构网络的联合表示
在得到药物和靶标的最终表示之后,前面提到的大多数基于相似度的方法都是将药物和靶标的表示串联起来,输入到神经网络中进行DTI预测。 然而,这种简单的组合策略忽略了药物与靶点之间的关键相关性。 受多模态表示学习(Cao et al., 2020; Guo et al., 2019; Tadas et al., 2019)的启发,我们提出了一种基于异构网络的联合表示框架来投影药物、目标、侧面的表示异质网络中的影响和疾病进入一个公共空间。 在异构网络中通过边连接的两个节点将在公共空间中更接近。 该方法考虑了多个模态之间的相关性以获得更好的嵌入表示,这对于后续的交互式学习过程尤为重要。
在公共空间中,药物、靶点、副作用和疾病的向量表示在模态相关的约束下被迫联系起来。 副作用和疾病没有生物学特征,所以我们首先假设副作用和疾病在公共空间的表示服从高斯分布: $\left{\begin{array}{l} E_{i}^{s e} \sim N\left(0, \sigma^{2}\right) \ E_{j}^{d s} \sim N\left(0, \sigma^{2}\right) \end{array}\right.$ 其中,$E^{se}_i$表示副作用 i 在公共空间中的可学习嵌入。$E^{ds}_j$表示疾病 j 在公共空间中的可学习嵌入。 在我们的实现中,$\sigma^2$被设置为 0.01。
为了将药物和靶序列的表示投影到公共空间中,我们使用了以下两个并行映射函数: $\left{\begin{array}{c} E_{i}^{d g}=f_{d g}\left(S_{i}^{d g}\right) \ E_{j}^{t g}=f_{t g}\left(S_{j}^{t g}\right) \end{array}\right.$ 其中,$S^{dg}_i$和$S^{tg}_j$分别表示药物i的序列表示和靶标j的序列表示。$E^{dg}i$和$E^{tg}j$分别表示药物和靶标在公共空间中的向量表示。$f{dg}(\cdot)$和 $f{tg}(\cdot)$是独立的映射函数,由具有多层感知器结构的神经网络近似。这里以药物为例,序列结构越相似,它们在公共空间中的距离就越近。
为了规范公共空间中副作用和疾病的可学习嵌入并规范可学习映射函数,我们提出了一种基于异构网络的联合表示框架。 基于异构网络的联合表示的目标函数如下: $L_{c} \mathrm{om}=\sum_{r \in R} \sum \begin{array}{l} M_{i}, N_{i}, \in r \ \left(M_{i}, N_{j} r\right) \in E \end{array}\left|E_{i}^{M}-E_{j}^{N}\right|^{2}$ 公式7 其中节点类型设置 $\mathrm{V}={\text { drug, target, disease, side effect}}$。 关系类型集$\mathrm{R}={ drug-drug-interaction, drug-protein-interaction, drug-disease-association,$ $drug-side \ effect-association, protein-protein-interaction, protein-disease-association }$。$E$表示异构网络中的边集。 $M_i$和 $N_j$分别代表类型 M 的节点 i 和类型 N 的节点 j。$E^M_i$和 $E^N_j$代表关系类型为$r$的两个节点 $M_i$和 $N_j$在公共空间中的嵌入。因此,公共空间中的药物嵌入、目标嵌入、副作用嵌入和疾病嵌入可以通过异构网络中的相关性相互增强,从而获得更好的表征。 值得一提的是,它们在公共空间中的嵌入是通过异构网络中生物序列和交互的表示获得的,适用于冷启动场景。
2.5 Prediction layer based on common space
如前所述,异构网络中由边连接的两个节点之间的距离在公共空间中会更近。 因此,我们设计了一个预测层,将公共空间中药物与目标之间的距离转换为 DTI 的预测分数。给定一个药物和靶标,他们在公共空间中的嵌入分别表示为$E^{dg}i$和$E^{tg}j$,那么他们之间的预测分数被表示如下: $x{i j}=\operatorname{Sigmoid}\left(B N\left(W E{i}^{d g}-E_{j}^{\operatorname{tg}} 2+b\right)\right)$ 其中,$W$ 和 $b$ 分别代表预测层的权重和偏差。$Sigmoid(\cdot)$ 表示 S 型饱和非线性激活函数。$BN(\cdot)$ 表示批量归一化操作。放在sigmoid函数前面,防止数据分布偏移进入饱和区,从而避免梯度消失。预测分数通过 sigmoid 函数正则化到 0 到 1 的范围内。
我们将预测 DTI 的任务视为一个二元分类问题,即药物与目标之间的相互作用被赋值为 0 或 1。我们使用二元交叉熵函数来拟合预测分数和标签值 . 目标函数如下: $\begin{aligned} L_{\text {pred }} &=-\sum_{(i, j) \in Z^{+}} \log {\mathrm{e}}\left(x{i j}\right)-\sum_{(i, j) \in Z^{-}} \log {\mathrm{e}}\left(1-x{i j}\right) \ &=-\sum_{(i, j) \in Z^{+} \cup Z^{-}} y_{i j} \log {e}\left(x{i j}\right)+\left(1-y_{i j}\right) \log {\mathrm{e}}\left(1-x{i j}\right) \end{aligned}$ 其中,$Z^+$ 表示观察到的药物和靶标相互作用的集合。 $Z^-$代表药物和靶标的负相互作用的集合。 $x_{ij}$和 $y_{ij}$分别表示药物 i 和目标 j 之间的预测分数和标签值。
为了对我们提出的“MultiDTI”中的参数进行正则化,我们最终通过联合正则化基于异构网络的联合表示和基于公共空间的预测层来定义我们的目标函数: $L_{\text {final }}=L_{\text {com }}+L_{\text {pred }}$
2.6 Training and testing
在异构网络数据集中,药物-靶标对的正样本比负样本少。 为了提高模型的泛化能力,我们将正样本过采样10次,对负样本进行欠采样,使大小与10次过采样后的正样本数相同。 之后,我们对采样数据进行 10 折交叉验证,即每折,随机选择 90% 的正负样本对作为训练数据,剩下的 10% 用于测试预测 模型的性能。 模型的最终性能取 10 倍结果的平均值。 在这里,我们使用小批量梯度下降来训练我们的模型,并使用反向传播策略来更新模型的相应参数。 具体来说,在每个 epoch 中,每批训练数据都采取梯度步骤来优化目标函数。 在我们的实现中,我们首先用几个批次优化 $L_{com}$,然后用几个批次优化$L_{pred}$。
为了防止我们模型中的神经网络过度拟合训练数据,我们使用了一种称为 Dropout 的方法。 这种方法在训练过程中停止了一些神经元的工作,只更新那些没有停止工作的神经元的参数。 在我们的实现中,dropout 用于方程 6 中的映射函数所在的神经网络。 这样,我们的模型将具有更好的泛化能力,并且可以扩展到测试场景。 另外,dropout只用在训练阶段,需要在测试阶段禁用。
2.7 Performance test metrics
在这项工作中,我们使用了六个性能指标:准确度、精确度、召回率、F1 分数、接收器操作特征曲线下面积(AUC)和精确召回曲线下面积(AUPRC)来评估模型。 AUC和AUPRC是根据曲线下面积得到的,其他四个指标定义如下: $\begin{array}{c} \text { Accuracy }=\frac{T P+F N}{T P+F P+T N+F N} \ \text { Precision }=\frac{T P}{T P+F P} \ \text { Recall }=\frac{T P}{T P+F N} \ \text { F1score }=\frac{2 * \text { Precision } * \text { Recall }}{\text { Rrecision }+\text { Recall }} \end{array}$ 其中,TP(True Positive)和TN(True Negative)分别代表正确预测的正负样本数。 FP(False Positive)和FN(False Negative)分别代表错误预测的正负样本的数量。 具体来说,如果某个药物-靶点对的预测得分大于 0.5,则模型确定药物和靶点相互作用,即为预测正样本,反之亦然。
3 Results
3.1 MultiDTI has superior predictive ability
我们将我们提出的模型“MultiDTI”与其他三个使用相似数据集的模型进行比较:“HNM”(Wenhui et al.,2014)、“DTINet”(Luo et al.,2017)和“NeoDTI”(Wan et al.,2019) )。 他们使用不同的方法来学习异构网络中节点的特征表示,然后根据特征表示进行 DTI 预测。 在数据集上,我们使用相同的异构网络数据集。 不同的是我们删除了原有异构网络(Wan et al.,2019)中的药物结构相似性网络和蛋白质序列相似性网络,使用序列特征提取器深度提取药物和靶标的序列特征。 异构网络。 值得注意的是,我们使用剩余的 10% 的部分异构网络数据集(第 2.1.1 节)除以 10 折交叉验证方法(第 2.6 节)来测试模型的性能。 从表 2 可以看出,我们提出的模型“MultiDTI”在 Recal、F1 分数、AUC 和 AUPRC 等指标中是最高的。 具体来说,‘HMN’模型的其他四个指标是没有的,我们在表格中用‘NA’来表示。 准确率是最常见的评价指标,即预测正确的样本数除以所有样本数。 在正负样本不平衡的情况下,准确率的评价指标有很大的缺陷。 “NeoDTI”表征异构网络并通过重构网络来训练模型。 用于训练和测试的负样本数量是正样本数量的十倍。 由于测试时负样本过多,‘NeoDTI’模型倾向于将样本预测为负样本,准确率值会更高。 精度表示归类为正例的样本实际上是正例的比例。 召回率表示所有实际为正的样本中预测为正的比例。 如果召回率相对较低,则意味着更多的正样本被模型预测为负样本。 F1分数综合衡量了模型的准确率和召回率。 它具有良好的声誉和客观性。 从这个指标来看,我们的模型优于其他模型,比第二高的模型高0.06。 同时,AUC 和 AUPRC 是常用的评价指标,用于测试二元分类器的能力。 我们的模型在这两个指标中也是最好的。 总的来说,‘MultiDTI’深度挖掘药物和靶点的序列信息,结合异构网络中的多重相关信息,具有强大的新DTI预测能力。
3.2 更多的相关信息和过采样可以提高预测性能
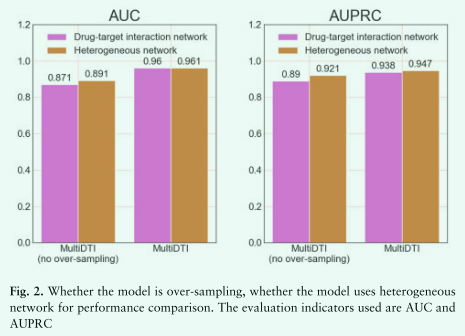
在这里,我们首先评估异构网络数据集对模型性能的影响。 在保持其他参数不变的情况下,我们只使用药物-靶点相互作用网络来重新训练模型并获得性能指标。 从图2可以看出,仅使用药物-靶点相互作用网络,模型的性能指标AUC和AUPRC都有不同程度的下降,对AUPRC指标的响应更加明显。 这些结果表明,异构网络中丰富的相关信息可以使药物和靶点在公共空间中得到更好的表达,从而提高预测 DTI 的性能。 另一方面,这也证明了我们提出的结合异构网络的相关信息和序列生物结构信息的联合表示学习框架的优越性。
在异构网络中,药物-靶标对的正负样本之间存在不平衡,正样本远少于负样本。 基于这个事实,当我们构建数据集时,我们将正样本过采样 10 倍,而对负样本进行欠采样。 欠采样后的负样本数与正采样后的正样本数相同。 在保证其他参数相同的情况下,我们对比了正样本过采样和没有正样本过采样的模型,结果如图2所示。我们发现正样本过采样10倍的模型有显着的提升 在 AUC 和 AUPRC 指标中。 这表明对正样本进行过采样(充分利用已知的药物-靶点对)可以增强模型的泛化能力,减少异构网络中数据不平衡的负面影响。
3.3 部分预测的新 DTI 得到 CHEMBL 数据库的确认
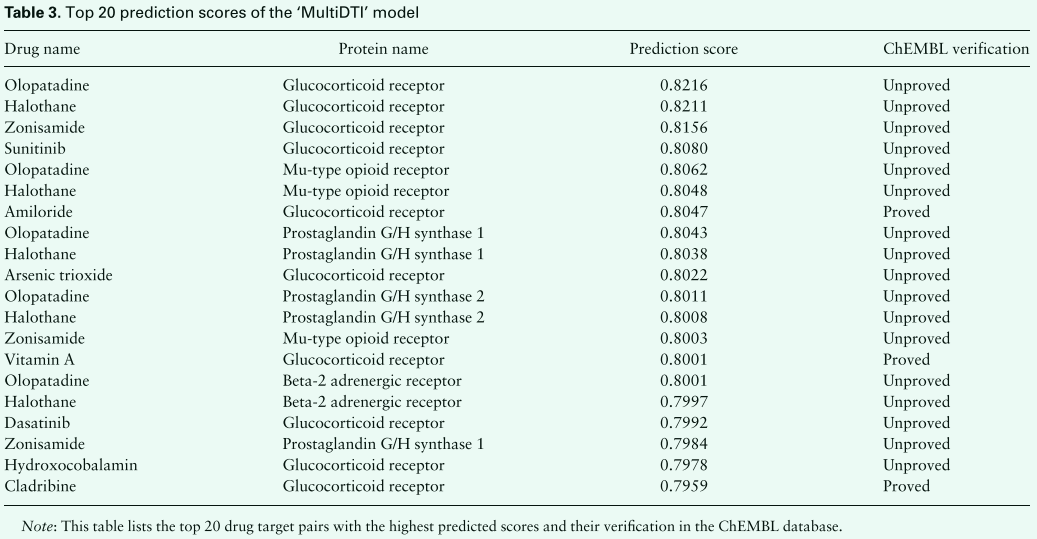
ChEMBL是一个具有生物活性的类药物小分子数据库,包含二维化学结构、计算特性和生物活性。 这些数据是从主要科学文献中提取和整理的,涵盖了大部分 SAR 和现代药物发现(Anna et al., 2017)。 我们使用这个数据库来验证我们的模型预测的新药物靶点对。 在这里,我们使用包含药物、靶点、副作用和疾病的完整异构网络作为数据集,并使用 10 倍过采样的正样本和相同数量的欠采样负样本来训练“MultiDTI”来预测新的 DTI。 通过10倍交叉验证的方法,我们得到了十个训练好的模型,我们将使用第一个训练好的模型来预测新的DTI。 我们将数据集中的所有药物和所有靶标(不包括在数据集中有相互作用的药物-靶标对)配对,将它们的序列输入模型并获得相应的预测分数。 我们将预测得分从大到小排序,并在表 3 中列出预测得分中排名前 20 的药物靶点(排名前 1000 的预测结果见补充数据)。 从表 3 中可以看出,ChEMBL 数据库已经验证了 3 个预测的药物-靶点对。 那些未经验证的药物靶点对是湿实验探索的良好候选者。 总体而言,这些由“MultiDTI”预测并由 ChEMBL 数据库确认的新 DTI 进一步证明了我们模型的强大预测能力。
3.4 新化学实体的目标预测
在这里,我们选择了一些新药来测试我们将新化学实体与已知 DTI 异构网络连接起来的模型的有效性。首先,我们将异构网络之外的药物输入到训练模型中。其次,该模型使用序列特征提取器根据其序列来表征药物。第三,模型使用多层感知器将药物的序列表示映射到训练好的公共空间,最后模型使用基于公共空间的预测层来预测药物的目标。在获得这些药物的预测靶点后,我们仍然使用ChEMBL数据库来验证预测结果。我们将序列长度更长的新药实体环孢菌素输入到模型中,经ChEMBL数据库验证的结果在预测得分前10位的靶点中,验证了9个靶点与环孢菌素相互作用。此外,我们将序列长度较短的新药实体格列本脲输入到模型中,得到的验证结果是,在预测得分排名前10位的靶点中,有9个靶点被验证与格列本脲相互作用。这些事实表明,我们的模型在新药实体的靶点预测方面具有很强的性能。
4 Conclusion
在这项工作中,我们提出了一种基于异构网络和序列的多模态 DTI 预测模型,名为“MultiDTI”。我们的模型在多模态表示学习中使用联合学习,将基于相似性的方法与基于网络的方法相结合。它不仅深入挖掘药物和靶点的结构信息,还考虑了异构网络中的交互或关联信息。我们的模型解决了大多数基于网络的方法只能预测网络数据集中的药物靶点的问题,并且我们的模型可以支持数据集外的药物-靶点对的预测。此外,我们的模型从 DrugBank 数据库中所有药物和靶点的序列中构建了一个序列片段词汇表,并将模型的固定序列长度设置为所有序列中最长的长度,可以支持最长序列的预测。药物-靶点对。与其他模型相比,我们的模型在实验中的预测性能有显着提升,一些预测的新 DTI 得到了证实。因此,我们认为‘MultiDTI’是预测新的DTI的强大而实用的工具,可以促进药物发现或药物重新定位的发展。