25 KiB
| title | date | updated | tags | categories | keywords | description | top_img | comments | cover | toc | toc_number | toc_style_simple | copyright | copyright_author | copyright_author_href | copyright_url | copyright_info | katex | highlight_shrink | aside |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Graph convolutional autoencoder and generative adversarial network-based method for predicting drug-target interactions | 2020-10-30 09:18:43 | 2021-03-23 09:41:06 | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | true | <nil> | <nil> |
摘要
对新型药物-靶点相互作用(DTI)的计算预测可以有效地加快药物重新定位的进程,降低药物重新定位的成本。以往的大多数方法都是通过构建浅层预测模型来综合药物与靶点的多种联系。这些方法没有深入学习药物和靶标的低维特征向量,忽略了这些特征向量的分布。提出了一种基于图卷积自动编码器和生成对抗网络(GAN)的DTI预测方法GANDTI。我们构建了一个药物-靶点异构网络,将药物与靶点之间的各种联系,即药物之间或靶点之间的相似性和相互作用,以及药物与靶点之间的相互作用整合在一起。建立了一个图形卷积自动编码器,用于学习药物和靶节点在低维特征空间中的网络嵌入情况,该自动编码器深度集成了网络中的各种连接。引入GaN将节点的特征向量归一化为高斯分布。在已知和未知的DTI之间存在严重的等级失衡。因此,我们构建了一个基于集成学习模型LightGBM的分类器来估计药物和靶的相互作用倾向。该分类器充分利用了所有未知的DTI,抵消了类不平衡带来的负面影响。实验结果表明,GANDTI预测DTI的性能优于几种最先进的DTI预测方法。此外,对五种药物的案例研究证明了GANDTI发现潜在药物靶点的能力。
Index Terms— adversarial regularization, drug-target interaction, generative adversarial network, graph convolutional autoencoder, LightGBM
介绍
药物通过药物-靶点相互作用(DTI)与各种分子靶点相互作用发挥药效。 蛋白质是这类分子靶标中的一组重要成员[1]。药物通过增强或抑制它们与之结合的目标蛋白的表达来影响疾病状态[2,3]。先前的研究已经证明,食品和药物管理局(FDA)批准的药物可以与几个靶点相互作用[4]。具有潜在未观察到靶点的现有药物可能具有未知的适应症[5]。然而,通过生物实验测定DTI是耗时、费力和昂贵的[6]。因此,许多研究已经开始通过计算方法预测新的DTI[7-9]。这些研究可以为生物学家提供DTI候选者,以减轻湿法实验室实验的工作量。
传统的DTI预测方法可以分为基于对接的方法和基于配体的方法[10]。基于对接的方法需要目标蛋白质的三维结构。由于不是所有目标的结构信息都是已知的,这些方法的性能受到限制[11,12]。基于配体的方法将具有未知配体的蛋白质与一组具有已知配体的蛋白质进行比较[13]。当已知配体的数量不足时,这些方法不能很好地执行。
近年来,许多研究已经开始从网络角度预测DTI[14,15]。这种方法通过整合异质药物靶点网络中的各种信息来分析潜在的DTI。 Chen等人。在药物靶标异质网络上应用随机游走来预测DTI[16]。Ezzat et al.。开发了一种基于图形正则化的矩阵分解模型来预测潜在的DTI[17]。这通过推断可能的DTI提高了药物-靶相互作用矩阵的密度,使得预测结果更加准确。 Luo等人。使用奇异值分解算法从药物和靶网络的邻接矩阵中提取有效信息[18]。这种基于矩阵分解的方法被称为DTINet。然而,随机游走和矩阵分解都是浅层模型,因此不能充分探索药物与其靶标之间的深层关系。
Bleakley和Yamanishi构建了一个二部局部模型,并用支持向量机(SVM)预测了DTI[19]。Lee和NaM提出了一种基于重启随机游走(RWR)和k近邻(KNN)的DTI预测方法[20]。他们将RWR应用于药物和目标网络。根据RWR结果对药物特征和目标特征赋予不同的权重。使用KNN模型计算每个药物-靶点对的相互作用分数。传统的机器学习模型如SVM、KNN通常只使用与已知DTI相同数量的未知DTI来训练模型。然而,两者之间存在着严重的阶级失衡。因此,这些模型的性能是有限的,因为大多数未知的DTI都被抛弃了。
为了预测新的DTI,Xuan等人。开发了一种称为DTIGBDT的集成学习方法,它可以用数据集中的所有样本训练模型[21]。该算法提取了基于路径类别的特征向量,融合了药物靶向异构网络的拓扑信息。 建立了基于梯度增强决策树的药物-靶点关联分析模型。同时,DTIGBDT没有深入学习药物和靶标的低维特征向量。
在这项研究中,我们开发了一种名为GANDTI的新方法来准确预测DTI。GANDTI通过图形卷积自动编码器深入集成了药物靶标异构网络的拓扑信息和节点属性[22]。编码器获得异构网络中药物节点和目标节点的嵌入表示。此外,通过生成性对抗网络[23]改变嵌入的表示以匹配高斯分布,这可以提高编码器[24]的稳健性。使用基于LightGBM的分类器计算DTI倾向[25]。LightGBM作为一种基于决策树[27]的集成学习[26]模型,能够充分利用未知DTI,有效地释放和消除类不平衡带来的负面影响。
材料和方法
我们的主要目标是通过分析药物和靶标属性,以及调查两者之间的相互作用来预测可能的DTIs。因此,我们构建了一个药物靶标异构网络,并提取了网络的边缘信息(网络拓扑)和节点信息(节点属性)。使用对抗性图卷积编码器学习网络中每个节点的特征表示。通过基于LightGBM的分类器计算药物-靶点对之间的相互作用分数。
数据集
DTI预测的数据集来自之前一项涉及549种药物和424个靶点的研究[18]。该数据集包括五种类型的数据:(1)549种药物的化学结构信息;(2)10,036种药物-药物相互作用(DDIS);(3)424个靶蛋白的初级序列;(4)7,363个靶-靶相互作用(TTI);以及(5)1,923个已知DTIs。蛋白质序列摘自人类蛋白质参考数据库(HPRD)[28]。其余的数据是从DrugBank数据库中获得的[29]。
药物靶向异构网络的构建
构建了DDI网络药网和TTI网络目标网。用D={d1,d2,...,dm}表示药物网络中的m个药物节点。DrugNet中的每条边表示由该边连接的两个药物节点之间的已知相互作用。类似地,我们使用T={t1,t2,...,tn}来表示targetNet中的n个目标节点。当两个目标节点具有已知交互时,添加边。此外,如果药物节点和目标节点之间存在已知的相互作用,则在这两个节点之间添加一条边。因此,基于数据集中的三种类型的相互作用(DDIs、TTIs和DTIS),我们构建了药物-靶点异构网络dtNet。
药物网、靶点网和异构的拓扑信息可以用这些网络的邻接矩阵来表示。例如,我们用$A^D$来表示药物网的邻接矩阵。当药物$d_i$和$d_j$之间存在边缘时,$A^D_{i,j}$=1,否则$A^D_{i,j}$=0。同样,目标网和数据网的邻接矩阵分别用$A^T$和$Y$表示。 我们根据药物的化学结构计算了药物之间的Jaccard相似系数(Jaccard similarity coefficients)[30],并构造了药物的相似矩阵,用$S^D$表示。利用目标的一级序列计算目标之间的史密斯-沃特曼(Smith-Waterman)评分[31],并构造相似度矩阵$S^T$。通过行归一化方法将$S^D$和$S^T$中元素的值缩放到[0,1],用来描述两个药物或靶点之间的相似性。一般认为,$S^D(i,j)$(或$S^T(i,j)$越接近1,$d_i$和$d_j$(或$t_i$和$t_j$)就越相似。
对抗性图形卷积自动编码器
我们的目标是确定dtNet中所有药物和目标的低维嵌入表示,例如药物和靶点特征向量。将特征向量送入分类器,计算药物与靶点之间的相互作用得分。
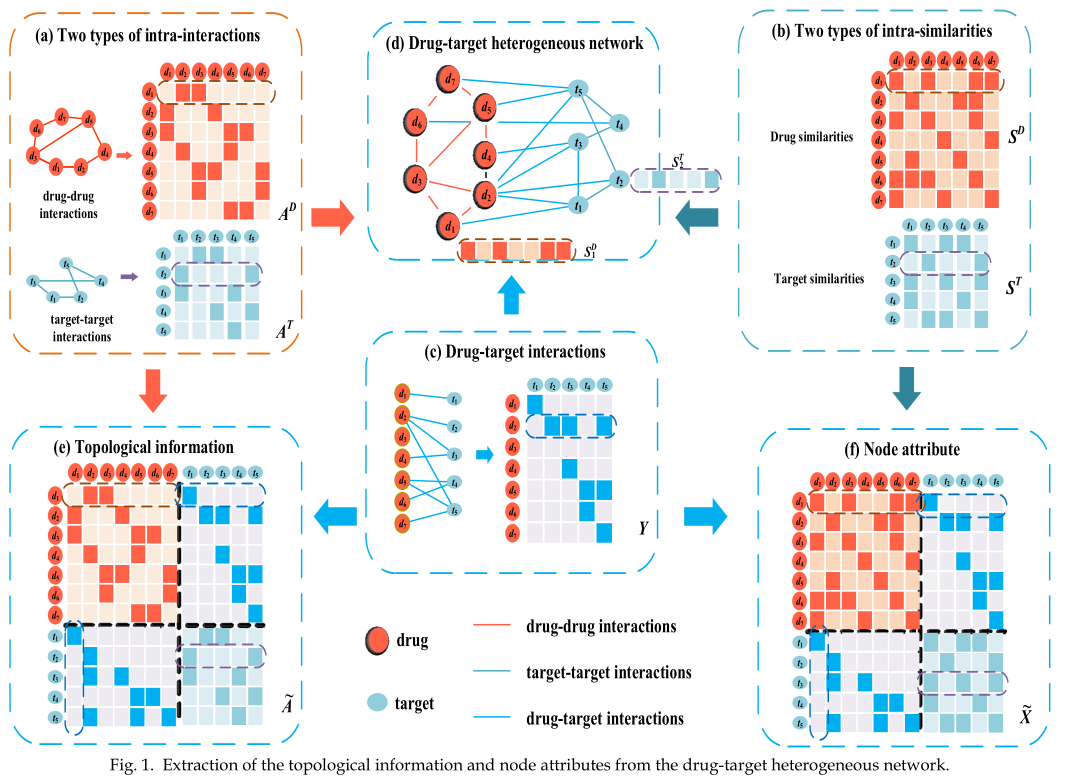 dtNet的拓扑信息矩阵由药物-药物相互作用矩阵$A^D$、靶-靶相互作用矩阵$A^T$和药物-靶相互作用矩阵$Y$组成。如图1所示,我们将这三个矩阵连接起来,得到了dtNet的拓扑关系矩阵$\tilde{A}$。
药物节点$d_i$的自身属性$\tilde{X_i}$由$S^D_i$和$Y_i$拼接而成,其中$S^D_i$是$d_i$与药物网络中其他药物的相似度向量,$Y_i$是$d_i$与dtNet中其他靶点之间的相互作用向量。类似地,靶点$t_j$的自身属性$\tilde{X_j}$通过连接相似向量$S^T_j$和相互作用向量$Y_j$来获得。
dtNet的拓扑信息矩阵由药物-药物相互作用矩阵$A^D$、靶-靶相互作用矩阵$A^T$和药物-靶相互作用矩阵$Y$组成。如图1所示,我们将这三个矩阵连接起来,得到了dtNet的拓扑关系矩阵$\tilde{A}$。
药物节点$d_i$的自身属性$\tilde{X_i}$由$S^D_i$和$Y_i$拼接而成,其中$S^D_i$是$d_i$与药物网络中其他药物的相似度向量,$Y_i$是$d_i$与dtNet中其他靶点之间的相互作用向量。类似地,靶点$t_j$的自身属性$\tilde{X_j}$通过连接相似向量$S^T_j$和相互作用向量$Y_j$来获得。
图形卷积编码器
 为了深入集成dtNet的拓扑信息以及药物节点和靶节点的属性,我们为我们的网络设计了一个基于图卷积网络(GCN)的编码器(图2(A))。该编码器有两个GCN层。为了充分利用药物靶标异构网络中各节点自身的信息,我们设置了$A'=\tilde{A}+I$。通过绘制拉普拉斯矩阵对拓扑信息矩阵$\tilde{A}$进行归一化,以获得$\bar{A}\in R^{(m+n) \times (m+n)}$,其中m是药物数量,n是靶点数量。可以计算如下:
为了深入集成dtNet的拓扑信息以及药物节点和靶节点的属性,我们为我们的网络设计了一个基于图卷积网络(GCN)的编码器(图2(A))。该编码器有两个GCN层。为了充分利用药物靶标异构网络中各节点自身的信息,我们设置了$A'=\tilde{A}+I$。通过绘制拉普拉斯矩阵对拓扑信息矩阵$\tilde{A}$进行归一化,以获得$\bar{A}\in R^{(m+n) \times (m+n)}$,其中m是药物数量,n是靶点数量。可以计算如下:
 其中$\tilde{D_{ij}}=\sum_j A'_{ij}$,$I$是单位矩阵。
如果要将dtNet中每个药物和靶点的特征向量投影到k维,则嵌入表示矩阵$Z\in R^{(m+n)\times k}$可以计算如下:
其中$\tilde{D_{ij}}=\sum_j A'_{ij}$,$I$是单位矩阵。
如果要将dtNet中每个药物和靶点的特征向量投影到k维,则嵌入表示矩阵$Z\in R^{(m+n)\times k}$可以计算如下:
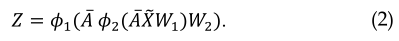 其中$W_1\in R^{(m+n)\times l}$和$W_2 \in R^{l\times k}$分别是第一和第二GCN层的权重矩阵。$l$是第一GCN层输出的特征图中每个药物和靶点的特征向量的维数。$\phi(\cdot)$是激活函数,$\phi_1(t)$=$Relu(t)$=$max(0,t)$,$\phi_2(t)=sigmoid(t)=\frac{1}{1+e^t}$。$Z$中元素的范围是$[0,1]$。矩阵$Z$的前m行表示m个药物的低维特征向量,后n行表示n个靶点的低维特征向量。因此,$Z$是深度融合拓扑信息和节点属性的低维表示。
其中$W_1\in R^{(m+n)\times l}$和$W_2 \in R^{l\times k}$分别是第一和第二GCN层的权重矩阵。$l$是第一GCN层输出的特征图中每个药物和靶点的特征向量的维数。$\phi(\cdot)$是激活函数,$\phi_1(t)$=$Relu(t)$=$max(0,t)$,$\phi_2(t)=sigmoid(t)=\frac{1}{1+e^t}$。$Z$中元素的范围是$[0,1]$。矩阵$Z$的前m行表示m个药物的低维特征向量,后n行表示n个靶点的低维特征向量。因此,$Z$是深度融合拓扑信息和节点属性的低维表示。
解码器和优化器
解码器的目的是利用嵌入表示矩阵$Z$重建dtNet的拓扑信息矩阵$\tilde{A}$。重构矩阵$\tilde{A}
\in R^{(m+n)
\times (m+n)}$中的元素$\tilde{A}(i,j)$,表示解码器预测的药物$d
_i$(或靶点$t_i$)与另一药物$d_j$或靶点$t_j$)之间的相互作用倾向。这可以通过公式3计算:
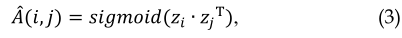 其中$z_i$和$z_j$分别是节点$i$和节点$j$的低维特征向量,${z_j}^T$是$z_j$的转置。
两个节点在低维特征空间中的特征分布越一致,两个节点对应的向量的内积越大,解码器的预测得分就越高。为了使拓扑信息矩阵$\tilde{A}$和结果矩阵$\hat{A}$尽可能一致,我们最小化了以下损失函数:
其中$z_i$和$z_j$分别是节点$i$和节点$j$的低维特征向量,${z_j}^T$是$z_j$的转置。
两个节点在低维特征空间中的特征分布越一致,两个节点对应的向量的内积越大,解码器的预测得分就越高。为了使拓扑信息矩阵$\tilde{A}$和结果矩阵$\hat{A}$尽可能一致,我们最小化了以下损失函数:
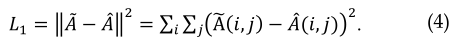 通过该图卷积自动编码器,我们深入挖掘了dtNet中节点之间的潜在关系。
通过该图卷积自动编码器,我们深入挖掘了dtNet中节点之间的潜在关系。
对抗性模型
假设药物或靶标的特征向量$z_i$服从高斯分布$z_i \sim q(z_i)$。为了提高编码器获得的特征向量的鲁棒性,我们引入了产生式对抗网络(GAN),使药物或靶标的低维特征向量更好地符合高斯分布。构造了一个基于多层感知器(MLP)的鉴别器D,以确定D的输入向量是来自发生器G还是来自高斯分布$z'_i \sim p(z'_i)$中采样得到的随机向量。
首先对高斯分布$z'_i \sim p(z'_i)$进行$m$+$n$次随机采样,得到真实样本集$Z'=\left{ z'_1,z'_2,z'3,...,z'{m+n} \right}$服从高斯分布,其中$z'i \in R^k$。为了避免鉴别器输入数据中的类别不平衡,我们在嵌入表示矩阵$Z$和真实样本集$Z'$中随机采样$a$个向量,以构造鉴别器D的输入矩阵$X^D \in R^{2a \times k}$。对于每个输入向量$x^D_i$,判别器给出0到1之间的分数,以确定该向量是来自$Z' \sim p(Z')$还是来自生成器G。分数越接近1,$x^D_i$越有可能以真正的高斯分布采样,反之亦然。通过鉴别器D获得的输入向量$x^D_i$的分数$s$可以计算如下:
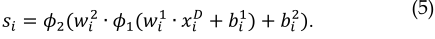 其中$w^1_i$和$b^1_i$分别是第一个全连接层的权重和偏置向量,$w^2_i$和$b^2_i$是第二个。$\phi_1(\cdot)$是$Relu$激活函数,$\phi_2(\cdot)$是$sigmoid$激活函数。GAN的$L_2$损失计算如下:
其中$w^1_i$和$b^1_i$分别是第一个全连接层的权重和偏置向量,$w^2_i$和$b^2_i$是第二个。$\phi_1(\cdot)$是$Relu$激活函数,$\phi_2(\cdot)$是$sigmoid$激活函数。GAN的$L_2$损失计算如下:
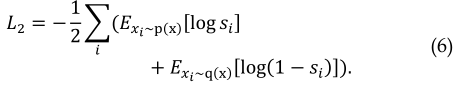 生成器G希望生成符合高斯分布的特征向量来愚弄鉴别器D,从而确保D给它打出尽可能高的分数。然而,鉴别器D的目的是对来自生成器G的输入向量进行尽可能低的评分,并对$p$中采样的输入向量进行尽可能高的评分。因此,GAN的优化目标可以定义为:
生成器G希望生成符合高斯分布的特征向量来愚弄鉴别器D,从而确保D给它打出尽可能高的分数。然而,鉴别器D的目的是对来自生成器G的输入向量进行尽可能低的评分,并对$p$中采样的输入向量进行尽可能高的评分。因此,GAN的优化目标可以定义为:
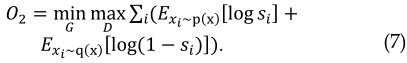
基于LightGBM的分类器
在我们的数据集中,已知的DTIs(阳性样本)和未知的DTIs(阴性样本)之间存在严重的类别不平衡。阳性和阴性样本的比例为1:120。对于训练传统的机器学习模型,如KNN [32]和SVM [33],使用了相同数量的负样本和正样本。许多含有有价值信息的阴性样本被放弃,限制了预测结果的准确性。 为了缓解类不平衡带来的负面影响,我们提出了一种基于LightGBM的集成学习模型作为分类器。LightGBM通过建立多个决策树,可以有效缓解类不平衡的影响。它使用不同的负样本数据集来训练不同的树,确保负样本的充分利用。
我们从对抗性图形卷积编码器接收到药物和靶标特征向量。如果使用$Z(d_i)$表示特征向量$d_i$,并使用$Z(t_j)$表示$t_j$的特征向量,则由$V_f(i,j)$表示的药物靶标对$(d_i,t_j)$的特征向量可以通过连接$Z(d_i)$和$Z(t_j)$来获得。假设数据集中正负样本的比例为$1:g$,建立$g$棵决策树,并用$T=\left{T_1,T_2,T_3,...,T_g \right}$表示。
对于前$k$棵决策树,我们可以从这些决策树中获得$k$个$V_f(i,j)$得分。基于这些得分和$V_f(i,j)$的标签,我们可以计算出残差$r_k$,它将被用作第$k+1$个决策树的标签。$r_k$可以计算如下:
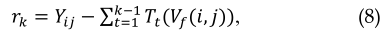 其中$T_t(V_f(i,j))$是第$t$个决策树的得分。所有阴性样本平均随机分为$g$组。
其中$T_t(V_f(i,j))$是第$t$个决策树的得分。所有阴性样本平均随机分为$g$组。
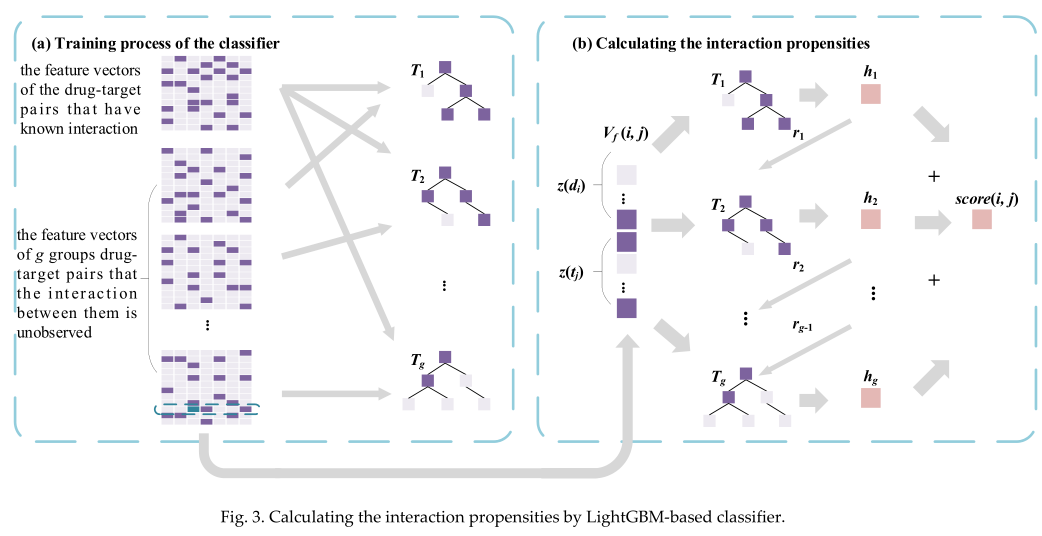 用一组负样本和所有正样本训练每个决策树$T_k
(1 \leq k \leq g)$(如图3(a)所示)。因此,在每个决策树的训练集中,都有相同数量的正样本和负样本。在对模型进行训练后,我们使用所有的决策树对$V_f(i,j)$进行评分,并将得分作为$d_i$与$t_j$有相互作用的倾向。$(d_i,t_j)$的相互作用得分可以定义如下:
用一组负样本和所有正样本训练每个决策树$T_k
(1 \leq k \leq g)$(如图3(a)所示)。因此,在每个决策树的训练集中,都有相同数量的正样本和负样本。在对模型进行训练后,我们使用所有的决策树对$V_f(i,j)$进行评分,并将得分作为$d_i$与$t_j$有相互作用的倾向。$(d_i,t_j)$的相互作用得分可以定义如下:
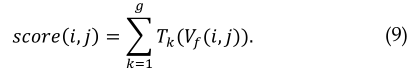 $score(i,j)$越高,$d_i$与$t_j$相互作用的可能性越高。相互作用得分矩阵$\hat{Y} \in R^{m \times n}$定义如下:
$score(i,j)$越高,$d_i$与$t_j$相互作用的可能性越高。相互作用得分矩阵$\hat{Y} \in R^{m \times n}$定义如下:
 用均方根误差评价GANDTI的损失。为了提高预测结果的准确性,阳性样本的预测分数被期望尽可能高,而阴性样本的预测得分尽可能接近0。因此,预测模型可以通过公式(11)进行优化:
用均方根误差评价GANDTI的损失。为了提高预测结果的准确性,阳性样本的预测分数被期望尽可能高,而阴性样本的预测得分尽可能接近0。因此,预测模型可以通过公式(11)进行优化:
 其中$Y_{ij}$是$d_i$和$t_j$之间真实的相互作用。
其中$Y_{ij}$是$d_i$和$t_j$之间真实的相互作用。
结果和讨论
绩效评估指标
五次交叉验证[34]被用来评估算法的性能。交叉验证的基本思想是将原始数据集分成不同的组。这些组依次作为训练集和测试集。
所有已知的DTI(阳性样本)被随机分成大小相等的5组。同样的操作也适用于未知的DTI(阴性样本)。在每一折验证中,四组已知和未知的DTIs用于训练模型,其余的DTI用于测试。因此,总共使用了1,536个已知DTIs和184,320个未知DTIs来训练模型。其余387个已知DTIs和46533个未知DTIs被视为测试集。在使用预测模型计算所有药物靶点的相互作用得分后,按其预测得分降序对样本进行排序。正样本的排序越高,该方法的性能越好。
对于给定的阈值$\delta$,如果阳性样本的预测得分大于$\delta$,则将其视为真阳性样本(TP)。如果阳性样本得分低于$\delta$,则定义为假阴性样本(FN)。如果阴性样本的得分大于$\delta$,则被认为是假阳性样本(FP)。如果不是,则视为真阴性样本(TN)。可以通过计算各种$\delta$下的真阳性率(TPRs)和假阳性率(FPRs)来构建接收器工作特性(ROC)曲线[35]。TPRs和FPRs可以定义如下:
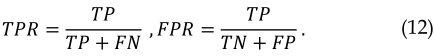 ROC曲线下面积(AUC)用于评估预测方法的性能[36]。普遍认为,AUC越接近1,该方法的性能越好。然而,先前的研究表明,对于类别不平衡的数据,P-R曲线下的面积(AUPR)是一个更具信息量的指标[37]。因此,我们还使用$AUPR$来评估我们的方法,该方法是通过精确度和召回率来计算的。查准率和召回率可以定义如下:
ROC曲线下面积(AUC)用于评估预测方法的性能[36]。普遍认为,AUC越接近1,该方法的性能越好。然而,先前的研究表明,对于类别不平衡的数据,P-R曲线下的面积(AUPR)是一个更具信息量的指标[37]。因此,我们还使用$AUPR$来评估我们的方法,该方法是通过精确度和召回率来计算的。查准率和召回率可以定义如下:
 此外,生物学家通常选择预测结果的顶部,通过wetlab实验进一步验证。因此,预测每种药物的前$k$个候选靶点的准确性更为重要[38]。因此,我们还显示了前$k(k=30,60...240)$个候选药物-靶点对的召回率,以揭示在前$k$个候选中有多少阳性样本被成功识别。
此外,生物学家通常选择预测结果的顶部,通过wetlab实验进一步验证。因此,预测每种药物的前$k$个候选靶点的准确性更为重要[38]。因此,我们还显示了前$k(k=30,60...240)$个候选药物-靶点对的召回率,以揭示在前$k$个候选中有多少阳性样本被成功识别。
参数设置
对于编码器的权重矩阵$W_1 \in R^{(m+n) \times l}$和$W_2 \in R^{l \times k}$,GANDTI中的设置为$l=500和k=200$。 GAN中的样本$a$的数量被设置为900。我们使用PyTorch在GPU(NVIDIA GeForce RTX 2070)设备上对神经网络进行了训练和优化。神经网络的$epoch$和学习率分别设置为2000和0.005。
与其他方法的比较
为了评估GANDTI的性能,我们将其与几种最先进的DTI预测方法进行了比较,包括DTIGBDT[21]、GRMF[17]、DTINet[18]和Lee的方法[20]。为公平起见,每个模型中的超参数均设置为相应文献的推荐值(DTIGBDT为$a=0.4, k=30, \lambda=0.1$;GRMF为$\eta=0.5, d=0.1, t=0.1,l=2$;DTINet为$r=0.8,\lambda=1$;Lee‘s方法为$r=0.8$)。特别地,我们将我们的模型与没有GAN的模型进行了比较,以证明GaN的效能。
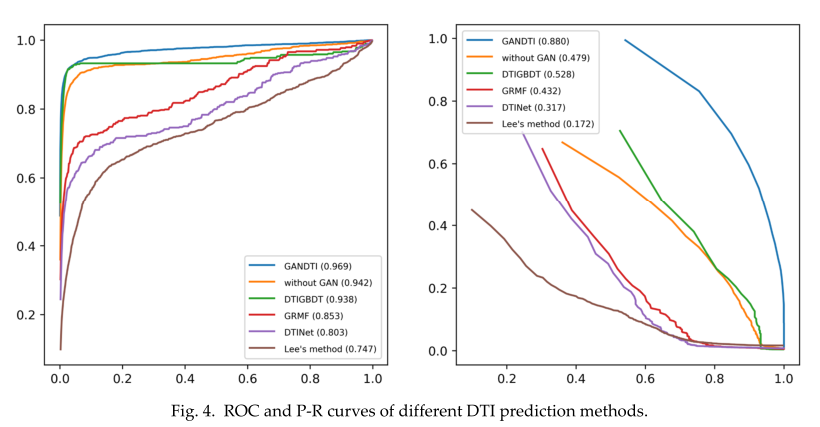 图4列出了每种方法的ROC和P-R曲线。GANDTI获得了最好的性能(AUC=0.969,AUPR=0.880),比第二种模型DTIGBDT获得了3.1%的AUC值和35.2%的AUPR值。DTIGBDT只提取了每个药物-靶点对的基于路径的特征,而没有学习它们更深层次的特征。与GRMF和DTINet相比,GANDTI的AUC分别比其他两种方法高11.6%和16.6%。GANDTI法比GRMF法高44.8%,比DTINet法高56.3%。这可能是由于异质药物靶标网络中的节点之间存在复杂的信息。基于矩阵分解的浅层预测模型GRMF和DTINet不能捕捉节点之间更深层次的潜在关联。Lee‘s法表现最差,AUC和AUPR分别比GANDTI法低22.2%和70.8%。由于KNN使用相同数量的负样本和正样本来训练模型,所以大多数负样本没有被利用。对于不含GaN的模型,GANDTI的AUC提高了2.7%,AUPRR提高了40.1%。结果表明,特征向量的分布影响预测结果的准确性。GANDTI的优异性能主要归功于该方法不仅集成了异构网络的拓扑信息和节点属性,而且充分利用了数据集的负样本。
图4列出了每种方法的ROC和P-R曲线。GANDTI获得了最好的性能(AUC=0.969,AUPR=0.880),比第二种模型DTIGBDT获得了3.1%的AUC值和35.2%的AUPR值。DTIGBDT只提取了每个药物-靶点对的基于路径的特征,而没有学习它们更深层次的特征。与GRMF和DTINet相比,GANDTI的AUC分别比其他两种方法高11.6%和16.6%。GANDTI法比GRMF法高44.8%,比DTINet法高56.3%。这可能是由于异质药物靶标网络中的节点之间存在复杂的信息。基于矩阵分解的浅层预测模型GRMF和DTINet不能捕捉节点之间更深层次的潜在关联。Lee‘s法表现最差,AUC和AUPR分别比GANDTI法低22.2%和70.8%。由于KNN使用相同数量的负样本和正样本来训练模型,所以大多数负样本没有被利用。对于不含GaN的模型,GANDTI的AUC提高了2.7%,AUPRR提高了40.1%。结果表明,特征向量的分布影响预测结果的准确性。GANDTI的优异性能主要归功于该方法不仅集成了异构网络的拓扑信息和节点属性,而且充分利用了数据集的负样本。
 为了验证GANDTI的AUC和AUPR值是否明显优于其他方法,我们进行了Wilcoxon检验[39]。表1中列出的统计结果显示,在p值阈值为0.05的情况下,GANDTI获得的性能明显优于所有其他方法。
为了验证GANDTI的AUC和AUPR值是否明显优于其他方法,我们进行了Wilcoxon检验[39]。表1中列出的统计结果显示,在p值阈值为0.05的情况下,GANDTI获得的性能明显优于所有其他方法。
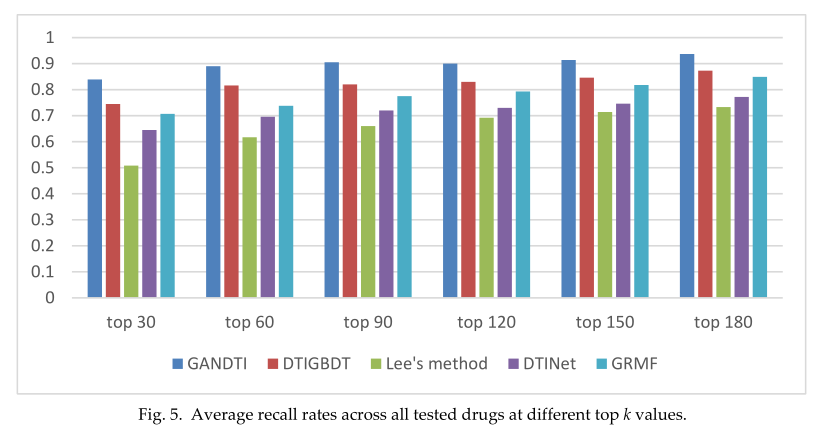 对于k个排序的目标,更高的召回率对应于更多的阳性样本被成功识别[40]。前k名中所有测试药物的平均召回率(k=30,60,90…180。对候选目标进行了计算,结果如图5所示。在不同的k值下,GANDTI的召回率优于所有其他方法。在前30名、前60名和前180名样本中,GANDTI的阳性率分别为83.9%、89.4%和93.7%。DTIGBDT获得了第二好的性能,在前30、60和180名中分别获得了74.5%、81.6%和89.3%的潜在DTI。GRMF在前30名中占70.7%,在前60名中占73.8%,在前180名中占84.9%。GRMF的性能略逊于DTIGBDT。DTINet在前30名中的查全率为64.5%,在前60名中的查全率为69.6%,在前180名中的查全率为77.2%,召回率高于Lee的方法。李的方法表现最差,在前30名中只有50.8%,在前60名中只有61.7%,在前180名中只有73.3%。
对于k个排序的目标,更高的召回率对应于更多的阳性样本被成功识别[40]。前k名中所有测试药物的平均召回率(k=30,60,90…180。对候选目标进行了计算,结果如图5所示。在不同的k值下,GANDTI的召回率优于所有其他方法。在前30名、前60名和前180名样本中,GANDTI的阳性率分别为83.9%、89.4%和93.7%。DTIGBDT获得了第二好的性能,在前30、60和180名中分别获得了74.5%、81.6%和89.3%的潜在DTI。GRMF在前30名中占70.7%,在前60名中占73.8%,在前180名中占84.9%。GRMF的性能略逊于DTIGBDT。DTINet在前30名中的查全率为64.5%,在前60名中的查全率为69.6%,在前180名中的查全率为77.2%,召回率高于Lee的方法。李的方法表现最差,在前30名中只有50.8%,在前60名中只有61.7%,在前180名中只有73.3%。
关于五种药物的案例研究
为了证明GANDTI发现潜在DTI的能力,我们对五种最相关的药物进行了案例研究,即奎硫平、氯氮平、奥氮平、阿立哌唑和齐拉西酮。表2列出了每种药物的前10个候选靶点。我们查阅了几个参考数据库和文献来源来支持GANDTI的预测结果。
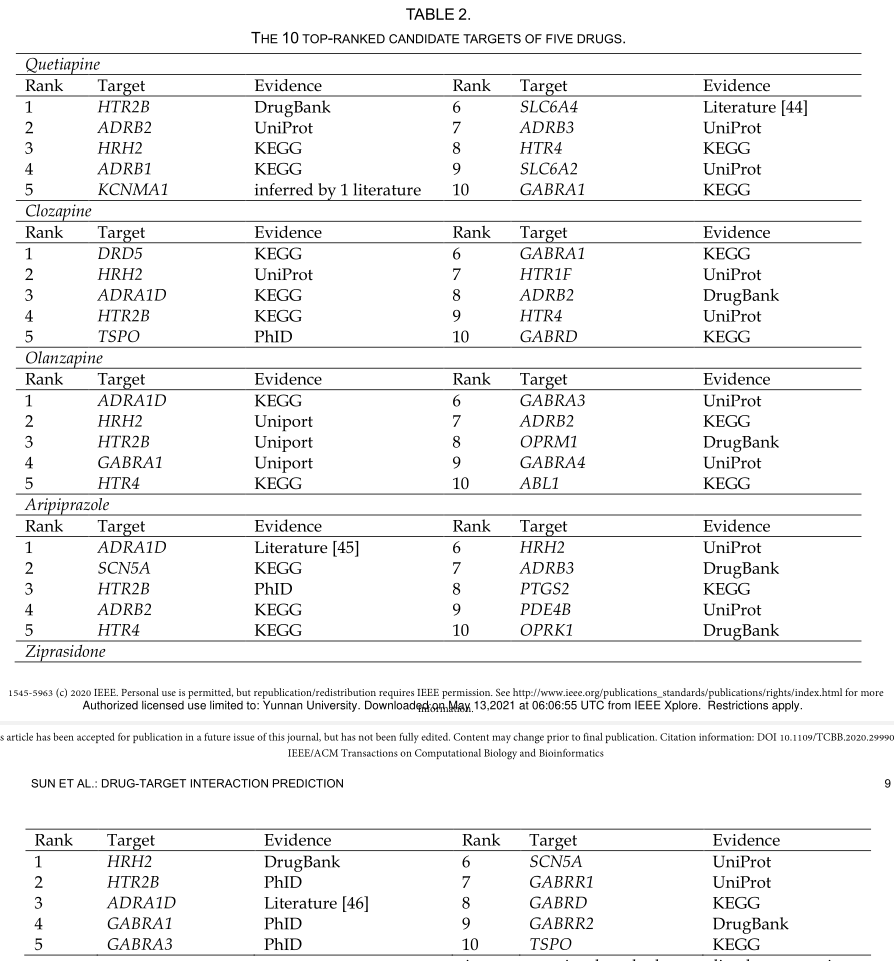 京都基因和基因组百科全书(KEGG)是一个用于检查药物-靶相互作用网络的数据库[41]。其数据主要来源于已发表的文献。另一个名为DrugBank的数据库提供了详细的药物数据,包括药物与靶标的相互作用[29]。Universal Protein (UniProt)是一个蛋白质数据库,其中包含大量来自文献的蛋白质生物学功能的信息,例如药物对蛋白质的调节作用[42]。如表2所示,KEGG数据库可以推断出19个候选DTIs,DrugBank可以推断出7个候选相互作用,UniProt可以推断出15个候选DTI,这意味着这些候选靶点可能会导致某种疾病,而这正是该药物的适应症。这些项目表明,这些药物可能会影响其候选靶点的表达。
京都基因和基因组百科全书(KEGG)是一个用于检查药物-靶相互作用网络的数据库[41]。其数据主要来源于已发表的文献。另一个名为DrugBank的数据库提供了详细的药物数据,包括药物与靶标的相互作用[29]。Universal Protein (UniProt)是一个蛋白质数据库,其中包含大量来自文献的蛋白质生物学功能的信息,例如药物对蛋白质的调节作用[42]。如表2所示,KEGG数据库可以推断出19个候选DTIs,DrugBank可以推断出7个候选相互作用,UniProt可以推断出15个候选DTI,这意味着这些候选靶点可能会导致某种疾病,而这正是该药物的适应症。这些项目表明,这些药物可能会影响其候选靶点的表达。
已经为网络药理学研究开发了一个名为PhID的数据库[43]。它包含了经过湿法实验室实验验证的真实药物靶标相互作用信息。一些标有“文学”标签的候选DTI得到了一些已发表文献的支持。表中5个候选DTI由PhID支持,3个候选被文献报道,表明这些药物与它们的候选靶点之间确实存在相互作用,并已得到实验证实。
除了手动验证的DTI之外,DrugBank数据库还包含一些从文献中推断的潜在交互作用。奎硫平的候选靶点KCNMA1位于DrugBank的推测部分,提示KCNMA1的表达可能受奎硫平的调节。表2中列出的所有候选DTI都得到了相关数据库或现有文献的支持。这表明了GANDTI确定潜在DTI的强大能力。补充表ST1列出了每种药物的30个高质量候选目标及其相互作用分数。
结论
开发了一种基于图形卷积自动编码器和生成式对抗网络的方法GANDTI来预测新的DTI。GANDTI的图形卷积自动编码器捕获了关于药物和靶的多种类型的内在联系,例如药物和靶的相互作用和相似性。同时,它还捕捉到了毒品和目标之间的相互联系(已知的DTI)。此外,药物和目标节点的低维特征分布被产生式对抗性网络规则化,用于增强DTI的证据。基于集成学习的分类器LightGBM充分利用了所有负样本,有效地抵消了类不平衡的影响。实验结果表明,GANDTI在AUC和AUPR方面的性能均优于本文测试的所有其他方法。对于生物学家来说,GANDTI是一种更有用的方法,因为它的顶级列表包含了更多实际的DTI。对五种药物的案例研究证明了GANDTI发现潜在DTI的能力。GANDTI是一个强大的优先排序工具,它为生物学家提供了可靠的候选DTI,用于随后通过湿实验室实验识别实际的DTI。
近年来,随着研究的深入,发现了一些可能影响基因表达和疾病进展的非编码RNA,如microRNA和长非编码RNA。一些研究还表明,非编码RNA可以被视为一种新型的药物靶点[47-51]。因此,我们的后续研究可能涉及到引入与非编码RNA相关的信息来辅助DTI的预测。