74 KiB
| title | date | updated | tags | categories | keywords | description | top_img | comments | cover | toc | toc_number | toc_style_simple | copyright | copyright_author | copyright_author_href | copyright_url | copyright_info | katex | highlight_shrink | aside |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Biomedical data and computational models for drug repositioning_ a comprehensive review | 2020-06-02 13:33:21 | 2021-07-20 20:31:38 | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | true | <nil> | <nil> |
摘要
药物重新定位可以显著降低传统药物研发的成本和持续时间,同时避免不可预见的不良事件的发生。随着高通量技术的快速发展以及各种生物数据和医学数据的激增,计算药物重新定位方法已经成为系统识别潜在药物-靶标相互作用和药物-疾病相互作用的有吸引力且强大的技术。在这篇综述中,我们首先总结了与药物、疾病和靶点相关的现有生物医学数据和公共数据库。然后,我们讨论了现有的药物重新定位方法,并根据它们的基本计算模型对它们进行分组,这些模型包括经典的机器学习、网络传播、矩阵分解和完成以及基于深度学习的模型。我们还全面分析了药物再定位中常用的标准数据集和评价指标,并对金标准数据集上的各种预测方法进行了简要比较。最后,我们总结了计算药物重新定位的挑战,包括降低生物医学数据的噪声和不完整性的问题,各种计算药物重新定位方法的集成,设计可靠的负样本选择方法的重要性,处理数据稀疏问题的新技术,大规模和全面的基准数据集的构建,以及对预测相互作用的潜在机制的分析和解释。 关键词:药物重新定位;药物目标预测;毒品疾病预测;计算模型;数据整合;评估标准
罗慧敏,中国湖南中南大学计算机科学与工程学院博士生,中国河南大学计算机与信息工程学院,开封,475001。她的研究兴趣包括生物信息学和计算药物重新定位。 李敏是中国湖南中南大学计算机科学与工程学院的教授。她的研究兴趣包括生物信息学和系统生物学。 杨梦云是中国湖南省中南大学计算机科学与工程学院的博士生。他的研究兴趣包括生物信息学和计算药物重新定位。 吴方翔是加拿大萨斯喀彻温省萨斯喀彻温省大学工程学院和计算机系的教授。他目前的研究兴趣包括生物信息学和人工智能。 李耀航是美国诺福克老自治领大学计算机科学系的副教授。他目前的研究兴趣是计算生物学、蒙特卡洛方法、大数据分析和并行/分布式/网格计算。 王建新是中国湖南中南大学计算机科学与工程学院的教授。他的研究兴趣包括计算基因组学和蛋白质组学。 提交日期:2019年10月23日;已收到(修订版):2019年12月7日作者2020年。牛津大学出版社出版。保留所有权利。如需许可,请发电子邮件至:journals.permissions@oup.com
Introduction
尽管在过去的几十年里,对药物研发的投入大幅增加,但批准上市的新药数量仍然很少。基因组学、生命科学和技术的最新进展并没有加快药物发现的进程。事实上,∼90%的候选药物在第一阶段临床试验中失败了,通常需要数十亿美元和10-15年的时间才能成功地将一种新药推向市场[1]。因此,探索提高药物研发成功率的有效方法迫在眉睫。近年来,药物重新定位已经成为药物发现中一个很有前途的领域,并引起了制药界和研究界越来越多的兴趣[2]。药物重新定位旨在为现有药物寻找新的治疗机会,以降低传统药物开发的时间、成本和风险,缩短药物审批和上市的周期[3,4]。药物重新定位的成功例子包括西地那非、沙利度胺和维甲酸。这些药物重新定位的成功案例进一步激励了全球制药行业探索现有药物空间的潜在容量。在过去的10年里,政府、学术研究人员和制药公司发起了大规模的资金和活动,以支持与药物重新定位相关的研究。国家先进转化科学中心已经启动了发现现有分子的新治疗用途项目[5]。加拿大卫生研究院已经建立了基金来支持药物再利用的基础科学研究[6]。
为了寻找现有药物的新适应症,制药行业通常可以采用两种策略,即基于活动的药物重新定位和计算药物重新定位[3,7]。作为一种实验策略,基于活性的药物重新定位是指在现有的一些全面的临床化合物数据库的基础上对药物进行检测。通过对数千种药物进行靶向或基于细胞的筛选,可以直接检测到药物的潜在适应症。虽然不需要目标或化合物的结构信息,但由于需要收集全部现有药物、专用设备和筛选分析,基于活性的方法仍然是一个费时费力的过程[8]。此外,在许多情况下,潜在的分子机制往往不清楚,这使得大规模重新定位药物变得困难。与基于活动的方法不同,计算性药物重新定位利用一些在线公开可用的数据库和生物信息学工具来检测药物、靶点和疾病之间的相互作用。它可以以更低的成本实现更快的重新定位过程,大多数制药公司最近已经采用了这种方法进行药物发现[7]。基本上,计算药物定位算法的快速发展和取得的成功可以归因于以下两个方面。首先,与药物相关的各个层面的大量高通量数据迅速积累,如基因组数据、蛋白质结构和表型。第二个方面是计算机科学的进步,这是开发有效的重新定位算法的基础[9,10]。计算方法有望针对不同的靶点和疾病有效地重新定位药物。
一般来说,传统的识别药物-靶标相互作用的计算方法主要有基于配体的方法和基于结构的方法[11-13]。基于配体的方法通过比较候选配体和已知的能与给定的目标蛋白结合的配体来预测与目标蛋白相互作用的药物。因此,当目标蛋白没有或只有很少的已知配体时,基于配体的方法可能效果不佳。基于结构的方法使用对接模拟技术来识别潜在的药物-目标相互作用(DTIs)基于已知的三维目标结构(3D)。这种方法计算量大,依赖于对接仿真方法的可靠性,不能应用于没有三维结构信息的目标。
机器学习技术通过探索药物-靶标和药物-疾病相互作用的全球模式,对基于配体和基于结构的方法进行了很好的补充,近年来已被广泛用于开发药物-靶标和药物-疾病预测的有效方法。基于机器学习的方法可以利用和整合来自药物发现研究的关于药物、靶点和疾病的异构多源生物医学数据,系统地识别潜在的药物靶点和药物-疾病相互作用。近年来,随着生物医学数据的增长,各种基于机器学习技术的计算药物重新定位方法被提出并成功应用。
基于机器学习的计算药物定位方法利用公开的数据库和生物信息学工具系统地识别药物、疾病和基因或蛋白质之间的相互作用。计算药物重新定位的工作流程如图1所示。首先,研究人员需要从与药物、靶点和疾病相关的各种公开可用的生物医学数据源收集可用的数据,这些数据可以为识别潜在的药物-靶点和药物疾病的相互作用提供有效的信息。其次,受益于计算机科学的进步,已经开发了各种模型来识别潜在的药物-靶点或药物-疾病相互作用。例如,纳波利塔诺等人。[14]利用已知的药物相关数据,包括基因表达、化学结构和靶信息来预测治疗类别。这些数据被用来计算三个单独的核。然后,通过对单个核函数的集成,定义了一个联合核函数,并将其作为支持向量机(SVM)分类的核函数。Li等人。[15]开发了一种新的基于相似性的方法,通过同类药物识别现有药物的新适应症。他们假设类似的药物有共同的适应症。根据药物化学结构和药物靶标信息计算两种药物之间的相似度。最后,为了评估和验证该方法的性能,需要在黄金标准数据集和其他测试数据集上与现有的方法在不同度量和预测任务下进行比较。
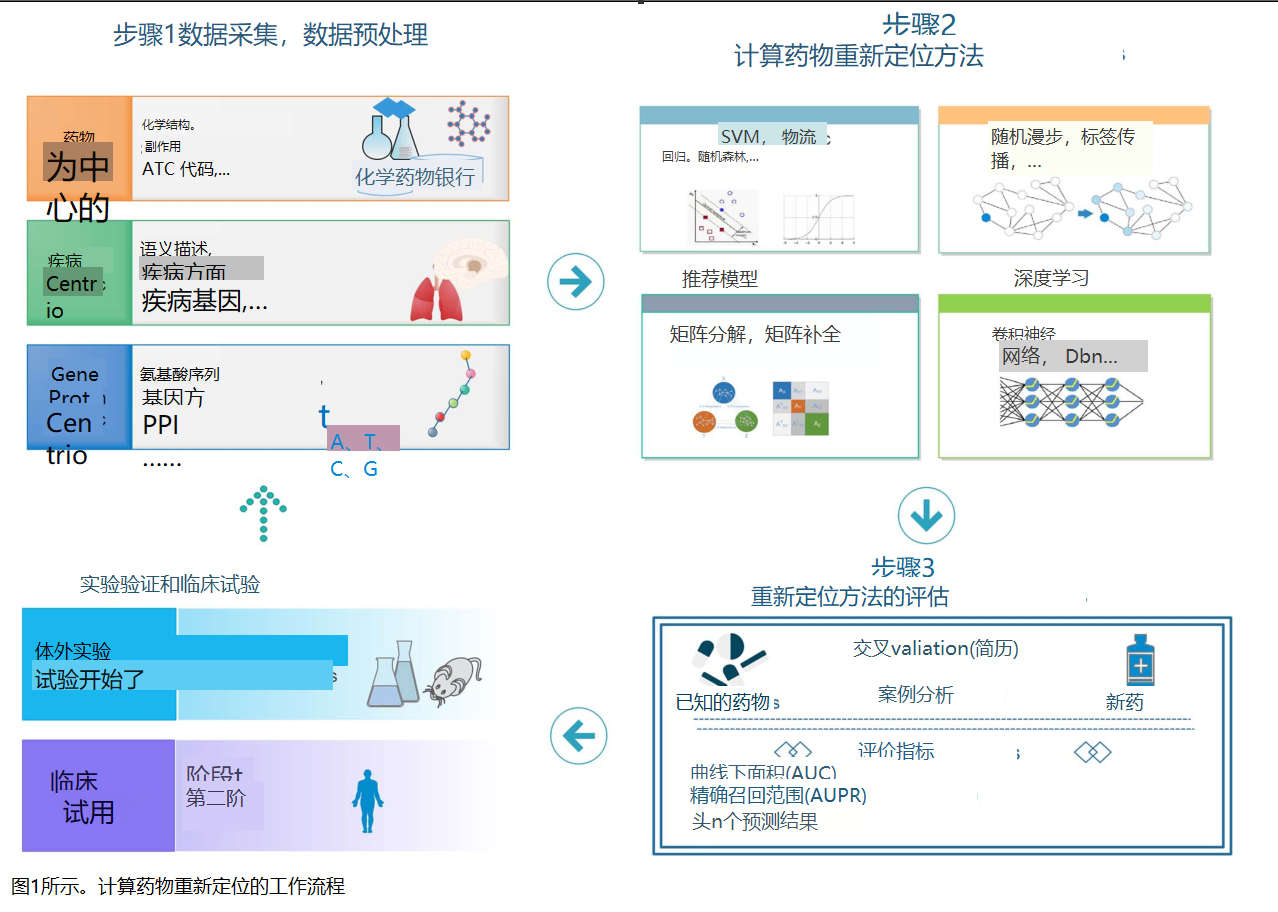
有关生物医学数据源的知识对于开发新的药物定位工具至关重要。由于现有数据的异质性和不完全性,理解各种生物医学实体之间的整体关系仍然是一个巨大的挑战[16]。尽管许多研究人员已经回顾了一些与药物定位相关的现有数据资源,但对于重要数据实体或它们与当前流行数据源之间的交互关系的全面描述,阻碍了研究人员快速、方便地获得所需的生物医学数据,以便开发高效的药物定位算法。更重要的是,虽然一些方法已被证明在解决药物重新定位问题上是成功的,但仍有一些悬而未决的挑战需要解决。首先,已知或验证的交互数据仍然不够。第二,使未知的相互作用成为真正的负值或在训练过程中忽略它们可能会降低计算模型对药物再利用的预测能力[17]。最后,对于分析和评价药物定位方法,金标准数据集和评价实验通常不一致。
近年来,从不同方面对计算药物定位的研究进展进行了详细的综述[1,18-24]。例如,Ding等人。[18]综述了用于DTIS预测的各种基于相似度的机器学习方法。朱等人。[19]回顾了现有的药物知识库及其在各种生物医学研究中的应用,包括药物重新定位。Ezzat等人的评论。[22]提供了计算性DTI预测方法的全面概述和实证比较。与以往的综述相比,本研究对基于机器学习的预测方法和相关生物医学数据在药物定位中的应用提供了更全面、更综合的分析。我们从讨论各种生物医学数据来源开始,这些数据来源使研究人员不仅能够理解和发现新的策略,而且还可以验证他们的结果,作为研究的一部分。其次,对目前流行的基于不同机器学习模型的药物定位方法进行了综述。最后,对药品再定位中使用的标准数据集和评价指标进行了综合分析,并在相同的金标准数据集上对各种方法进行了简要的比较,评价了各种方法的优势和局限性。
在周等人最近的评论中。[25]详细讨论了DTI识别的相关数据库和不同的计算模型。在这篇综述中,我们主要从机器学习的角度对现有的潜在药物靶点或药物疾病相互作用预测的计算方法进行分析和分类。虽然我们调查中涉及的方法与[25]有一定的重叠,但我们用基于矩阵补全的计算方法对其进行了补充,这些方法最近在药物重新定位中显示出有吸引力的预测精度增强。我们进一步讨论了各种预测模型的优缺点以及与之相关的问题,如冷启动、稀疏性和噪声数据问题。此外,我们的调查扩展了与不同生物医学实体相关的各种类型的数据,包括药物、疾病、基因和蛋白质。还包括了用于药物重新定位研究的通用验证策略、评估指标和基准数据集,以指导研究人员在未来的研究中有效地评估和验证他们开发的方法的预测能力。
可用的生物医学数据
高通量生物学的最新技术进步已经产生了海量的多组学数据,如基因组、蛋白质组和代谢组学数据,以及来自药物基因组学、临床和化学来源的其他数据。这些数据大多存储在数据库中,这些数据库可供公众进一步研究和分析。在某种程度上,它们为开发高效的药物重新定位方法和工具创造了前所未有的机遇。为了计算药物的重新定位,研究人员通常需要收集或融合从多个数据源获取数据,构建包含更多隐藏信息的生物医学网络。然而,其中一个主要困难是如何收集和分析所需的生物医学数据,因为它们是异构的,不同实验产生的数据包括不同类型的信息,如核苷酸序列和蛋白质-蛋白质相互作用。此外,个别小组或研究机构提供的数据集在实体注释和数据格式方面往往表现出不一致和脱节,不能用来直接与其他数据库链接。因此,全面了解这些可用的数据库可以缓解实体识别和数据不一致带来的问题。此外,它还可以帮助提高数据的准确性,并加快后续的数据分析过程。本节的目的是回顾与不同生物医学实体相关的各种类型的数据,包括药物、疾病、基因和蛋白质,这些数据可以帮助从不同来源提取生物医学信息。补充表1总结了这些常用数据源[26-59]的列表,其中提供了简要说明和访问链接。根据涉及的生物医学实体,这些流行的公共数据源可分为四类:药物中心数据库、疾病中心数据库、基因/蛋白质中心数据库和综合数据库。
以药物为中心的数据
在过去的几十年里,制药工业和学术药物发现工作取得了稳步进展,如化合物合成和分析、化学生物学或基因组学,这些工作产生了关于新化合物及其相应生物活性的有价值的报道。在一些流行的药物化学期刊上,每年大约有20000-30000个新化合物发表,而且这个速度在最近几年一直在加速[60]。在此基础上,一些研究机构收集了有关药物或类药物化合物及其与人体生物系统相互作用的现有信息,称为以药物为中心的数据,并构建了许多有价值的数据库。如表1所示,有些药物数据库是免费提供和普遍使用的,可以用来建立基于知识的模型,使我们能够进行分析和预测。表1中描述的这11个以药物为中心的数据库存在显著差异,这是由不同的生物医学数据源和数据类型引起的。对于研究人员来说,了解如何在每个以药物为中心的数据库中描述和组织药物或类药物化合物是至关重要的。我们访问了这些数据库,一步一步地浏览和分析了它们的内容。在这里,我们的目的是通过讨论一些用于药物重新定位的流行资源来帮助研究人员理解和利用这些以药物为中心的数据库。它概述了表1中的主要内容。
 通常,药物或类药物化合物的性质从以下几个方面描述或量化。首先,可以根据药物的物理和化学性质对其进行识别和定量。例如,包括DrugBank和SuperTarget在内的一些数据库提供了分子结构,可用于一些计算化学相似性的方法或工具箱,如CDK[61]和SIMCOMP[62]。第二,分子活性和表型对相关药物的分析非常有效。药物或类药物化合物通过与细胞蛋白结合来发挥其生物活性。一些研究人员利用药物与靶标或蛋白质之间的交互特征信息来计算药物之间的相似性[63]。疾病和副作用也可以用来量化和表征药物或类药物化合物[64]。当两种或两种以上药物一起服用时,药物与药物的相互作用可能发生,并能反映药物之间的相似性[65]。
通常,药物或类药物化合物的性质从以下几个方面描述或量化。首先,可以根据药物的物理和化学性质对其进行识别和定量。例如,包括DrugBank和SuperTarget在内的一些数据库提供了分子结构,可用于一些计算化学相似性的方法或工具箱,如CDK[61]和SIMCOMP[62]。第二,分子活性和表型对相关药物的分析非常有效。药物或类药物化合物通过与细胞蛋白结合来发挥其生物活性。一些研究人员利用药物与靶标或蛋白质之间的交互特征信息来计算药物之间的相似性[63]。疾病和副作用也可以用来量化和表征药物或类药物化合物[64]。当两种或两种以上药物一起服用时,药物与药物的相互作用可能发生,并能反映药物之间的相似性[65]。
为了药物定位的目的,我们可以选择一些流行的数据库,如DrugBank,以获得关于分子结构或交互图谱的基本生物学数据。有时,研究人员希望整合来自多个以药物为中心的数据库的数据,以获得药物或类药物化合物及其相互作用信息的综合视图。在这种情况下,需要解决几个实际问题。首先,在以药物为中心的数据库中,一种药物通常存在许多不同的名称和标识符。因此,在集成过程中可能会出现一些映射错误或数据丢失。因此,需要一个标准化的数据集成命名或映射规则,以获得更好的性能,避免集成实验数据集的不一致。幸运的是,一些研究人员已经开始关注这个问题,并提出了初步的解决方案,例如RxNorm[66]。其次,资源提供方式的差异也是理解和整合以药物为中心的数据库的一个挑战问题。例如,化学结构的格式是不同的,可以用简化的分子输入行输入系统(SMILES)、MOL、SDF、PDB或其他格式来表示。目前,针对这一问题,研究人员可以利用一些开放的软件系统将一种文件格式转换成另一种格式。在未来,所有以药物为中心的数据库都有望为数据集成提供标准的、统一的分子结构格式。第三,一些数据库不提供可下载的完整数据集,研究人员只能搜索相关数据来构建他们的数据集。同样,以药物为中心的数据库内容的差异和数据组织过程的不一致导致了用于药物重新定位的金标准数据集的偏差。
以疾病为中心的数据
以疾病为中心的数据库使研究人员能够访问各种与疾病相关的数据,并在生物信息学中发挥核心作用。在过去的几十年里,人们研究了许多与疾病相关的分子遗传学、表型和药物,同时开发了与疾病相关的数据库来了解疾病的本质。例如,在线人类孟德尔遗传数据库(OMIM)是疾病遗传信息的主要储存库。
在以前的研究中,疾病根据不同的标准进行分类,如病理学、解剖学、预后、分子遗传学和药物[67]。在这里,我们的目标是关注流行的以疾病为中心的数据库中一些最相关的特征,以帮助研究人员理解和使用这些数据库。通过浏览和分析这些数据库中的信息,与疾病相关的数据根据药物重新定位应用被分成四个有价值的类别,如表2所示。
语义结构常被用来计算疾病之间的相似度,这是研究疾病分子机制的一个重要因素。此外,疾病术语通常提供一些关于疾病表型的关键信息,一些以疾病为中心的数据库对人类疾病提供了明确的定义。研究人员一般从疾病数据库UMLS、DO、MESH、HPO等中获取结构信息或疾病术语,还可以从OMIM、DisGeNET等数据库中获取与特定疾病相关的基因。遗传信息可以帮助研究人员在分子水平上了解疾病。相比较而言,除了MalaCard和DiseaseConnect之外,这些数据库不提供与疾病相关的这种药物信息。
一般来说,这些数据库是研究人员进行研究的宝贵资源。疾病标注或本体结构的质量对药物重新定位的准确性有很大影响。此外,这些信息源普遍存在术语的异构性和内容的不完备性,严重阻碍了数据的共享和交换。DO数据库对生物医学数据库中人类疾病标注的标准化起着重要作用。DO数据库在语义上集成了多种疾病和医学词汇,并提供了这些词汇之间的交叉引用。因此,很多研究者总是把疾病术语统一起来做。我们期待疾病本体论的进一步发展,以便将基因和基因组数据与人类疾病联系起来。
基因/蛋白质中心数据
基因和蛋白质为合理的药物设计提供了基础。最近,许多以基因和蛋白质为中心的数据库已经成功开发,如表3所示。通常,氨基酸序列、基因术语和蛋白质-蛋白质相互作用通常包含在以基因/蛋白质为中心的数据库中。氨基酸序列是指按特定顺序构成蛋白质分子的一串氨基酸。对于药物重新定位,UniProtKB和HPRD数据库提供蛋白质的氨基酸序列信息,它可以用来计算目标之间的相似度和表示目标特征。同样,基因术语和蛋白质-蛋白质相互作用也可以用来确定药物的潜在靶点。此外,一些研究人员利用蛋白质-蛋白质相互作用来构建靶点网络,以寻找潜在的药物-靶点对。蛋白质的三维结构也可以为理解其在疾病中的作用提供重要信息。

集成数据
上述研究主要收集一类生物医学实体的相关数据。然而,单一数据源可能是不完整的或有限的。因此,在实践中集成来自多个来源的各种生物医学数据是非常重要的。有研究表明,数据整合有助于提高药品重新定位的性能。例如,Wang等人。[68]根据化学结构、靶蛋白和多种药物的副作用分别计算药物相似度消息来源。然而,在大多数情况下,研究人员需要从多个数据库中浏览所需的数据并手动整合,这减缓了药物重新定位方法的快速发展。综合数据库可以有效地辅助药物定位的研究。逐步建立了包含药物、疾病、基因、途径和各种相互作用信息的综合数据库,如补充表1所示。
在最近的研究中,大多数药物定位方法都是利用药物、疾病和目标的相似性或特征数据来实现的。基于上述数据分析,可以利用从不同方面和不同层次描述药物、疾病和目标的多个数据来衡量相似性或构建特征。生物医学实体的相似性和特征信息可以进一步合并到不同的预测模型中,以识别新的药物相关关联。
计算型药物定位方法
各种机器学习技术已经被用来开发有效的药物定位预测方法。根据应用于计算药物定位方法的机器学习模型,这些药物定位方法主要分为四类:经典机器学习模型、网络传播模型、矩阵分解和完成模型以及深度学习,如图2所示。

基于经典机器学习模型的方法
近年来,快速发展的机器学习技术为药物重新定位提供了有效的方法,包括DTI和药物-疾病相互作用预测。这些方法的动机是观察到类似的药物往往针对类似的蛋白质,反之亦然[69],或者类似的药物往往针对类似的疾病,反之亦然[70]。一个直观的想法是将药物重新定位制定为一个二元分类问题,其中药物-靶点和药物-疾病对作为实例处理; 将药物、疾病和目标的信息作为特征处理。然后,可以使用经典的分类模型,如支持向量机(svm)、正则化最小二乘(RLS)、逻辑回归(logistic regression)和随机森林(random forest)来开发药物重新定位方法。
支持向量机 Bleakley等[71]提出了一种新的监督推理方法,用支持向量机作为局部分类器的二部局部模型(bipartite local model, BLM)来预测新的DTIs。对于候选药物-靶标对,可以结合两个独立的预测,包括预测所给药物的靶蛋白和预测针对所给蛋白质的药物,从而对每个相互作用给出明确的预测。BLM在涉及酶、离子通道、G蛋白偶联受体(GPCRs)和人类核受体的四个金标准数据集上取得了更好的性能。然而,BLM有其局限性,因为它不能在没有任何相互作用的情况下对新药/靶标候选物提供合理的预测,这被称为冷启动问题。
考虑到多种类型的数据源可以有效集成以提高预测性能,Wang等[68]提出了一种新的药物重新定位方法PreDR (Predict drug repositioning),该方法将用于定义药物与疾病相似度的分子结构、分子活性和表型数据整合在一起。特别地,药物和疾病用它们的相似谱来表示,并构造核函数来计算药物-疾病对之间的相似度。然后,训练具有定义核的支持向量机来发现新的药物-疾病相互作用。Wang等[72]利用数据融合技术收集药物药理和治疗效果、药物化学结构和蛋白质组学信息来衡量药物相似度和目标相似度,并在基于svm的预测器内通过一个高效的核函数来整合这些相似度。
大多数现有的机器学习方法使用实验验证的交互作用作为未验证交互作用的正样本和负样本来训练预测模型[73]。 然而,随机生成的负样本可能包含未知的真实阳性样本,这可能导致决策边界存在偏倚[74]。因此,在基于监督学习的药物重新定位方法中,筛选高可靠的阴性(RN)样本是很重要的。基于这种挑战,Liu等人[73]提出了一种识别高RN样本的系统筛选流程。筛选框架的建立是基于这样一种假设:与已知/预测的特定化合物目标不同的蛋白质不太可能被化合物靶向,反之亦然。进行了大量的实验,结果表明,筛选的阴性样本可以有效地提高预测方法的准确性。
DTI识别的困难包括已知DTI的稀疏性和没有实验验证的阴性样品。Lan等[75]提出了一种DTI预测方法PUDT。他们把已知的相互作用设为正样本,把未知的相互作用设为未标记的样本。根据目标相似度,将未标记的样本分为RN样本和可能的阴性样本,分别采用重启随机漫步、KNN和热核扩散三种策略。然后,将三种策略的结果汇总,确定未标记样本的最终标签。最后,提出了一种基于加权支持向量机的多级分类器来识别潜在的DTIs。
Peng等[76]开发了一种基于阳性无标记学习的阴性样本提取方法,用于从未知DTIs中识别阳性、阴性和模糊DTI样本。然后,计算了属于正类和负类的模糊样本的概率。通过整合筛选的样本,构建基于svm的优化模型来预测新的DTIs。
正则化最小二乘 Xia等[77]在Laplacian RLS方法的基础上,提出了一种半监督学习方法NetLapRLS,该方法整合了化学空间、基因组空间和药物-蛋白质相互作用网络空间的信息,预测药物-蛋白质相互作用。通过纳入经过验证的药物-蛋白质相互作用信息,标准LapRLS得到了改进。
Van Laarhoven等人[78]提出了一种简单的RLS算法,结合了由DTI配置文件构建的内核产品。将所构建的高斯相互作用谱(GIP)核与化学信息和基因组信息相结合,引入了基于Kronecker产品的药物核和目标核相结合的RLS-Kron算法,取得了良好的性能。Nascimento等[79]对KronRLS方法进行了扩展,提出了一种新的预测算法KronRLSMKL,该算法利用多核学习算法在两部分药物蛋白预测问题上自动选择并结合核。核可以看作是在所有实例对上估计的相似矩阵。Laarhoven等人[80]将一种简单的加权最近邻方法集成到RLS-Kron分类器中,用于DTIs的预测。具体来说,加权最近邻法通过使用训练集中所有药物的相互作用轮廓和相似信息来定义一个新药物化合物的轮廓。
许多基于核的预测方法是将各种核线性组合来构造核矩阵,当核之间存在非线性关系时,这种方法并不适用。因此,Hao等[81]将RLS与非线性核融合算法相结合,提出了一种DTI预测方法RLS- kf。采用核融合技术将相似矩阵与高斯核矩阵相结合。Li等[82]提出了一种新的灵活、鲁棒的多源学习(FRMSL)框架,利用Kronecker RLS (KronRLS)方法集成不同来源的生物数据来测量药物相似度和疾病相似度,并解决预测问题。
逻辑回归和核回归 Perlman等[63]结合多种药物-药物和基因相似性措施,开发了基于相似性的DTIs推断框架。基于给定的一对药物-药物和基因-基因相似度量,计算每个药物-靶标对的关联得分。然后,一个整合多个测量值的逻辑回归分类器被训练来预测新的DTIs。
基于观察到类似的药物表示类似的疾病,Gottlieb等。[83]提出了一个计算方法预测,构造多个药物之间和disease-disease相似的措施,遵循[63]的方法构造分类功能,后来学了逻辑回归分类器预测小说药物和疾病之间的联系。
Yamanishi等[84]提出了一种基于核回归的方法来预测新的DTIs。首先,根据药物-靶标二部图将药物和靶标嵌入到统一的药理学空间中。然后,学习药理空间与化学/基因组空间相关性的核回归模型,推断新化合物/蛋白质的药理特征。化合物和蛋白质之间的紧密程度是根据它们的药理特征来测定的。Yamanishi等[85]通过研究化学空间、药理空间和DTI网络拓扑结构之间的关系,发现药理相似度比化学结构相似度更有助于预测DTIs。因此,他们提出采用[84]中提出的利用药理学空间和基因组空间的学习方法来推断DTIs。
随机森林 Olayan等[86]开发了一种高效的药物靶标预测计算方法,称为DDR。通过使用各种药物和靶标相关数据计算出药物和靶标的多个相似度量。然后,对这些相似测度进行选择和融合,构建复合相似度。通过整合药物相似性、目标相似性和已知DTI,构建DTI异质图。DDR从构建的DTI异构图中提取基于图的特征,训练随机森林模型预测DTIs。曹等。[87]提出了一种结合化学、生物和DTI网络信息的DTI预测方法。一个药物靶标对可以用药物的化学信息、靶标的结构信息和相互作用信息三种特征来表示。然后,利用综合特征的随机森林模型建立DTI预测方法。
基于网络传播的方法
除了学习分类器来预测DTI和药物与疾病的相互作用外,基于网络的方法也得到了广泛的应用[88]。它的目的是利用网络来组织生物和生物医学实体之间的关系,以确定网络层面上潜在的新型关系[16]。基于网络的分析已经成为计算药物重新定位的一种广泛使用的策略。近年来,许多基于网络的方法被提出,并成为流行的药物重新定位工具。
随机游走 Chen等人。[89]假设相似药物与相似靶点相互作用,提出了一种基于网络的异质网络上重新启动的随机游走(NRWRH)方法。基于已知的DTI,NRWRH构造了一个新的药物相似度矩阵和一个新的靶点相似度矩阵。将药物化学相似度矩阵和新药相似度矩阵合并为一个综合药物综合相似度矩阵,将靶蛋白序列相似度矩阵和新靶点相似度矩阵合并为一个综合靶点相似度矩阵。然后,将两个集成的相似度网络和已知的DTI数据进行集成,构建了药物-靶点异构网络。最后,在构建的异构网络上实现随机游走算法,预测药物与目标的相互作用概率。刘等人。[74]提出了在药物-疾病异质网络TP-NRWRH上重新启动的两遍随机游走算法,以识别潜在的药物-疾病关联。首先,构建了由药物相似网络、疾病相似网络和已知药物疾病关联网络组成的异构网络。然后,他们扩展了NRWRH,实现了两次随机行走并重新开始,包括以药物为中心的随机行走和以疾病为中心的随机行走,以确定未知药物-疾病对的相互作用似然值。
罗等人。[90]利用已知的药物-疾病关联来改进药物-药物和疾病-疾病的相似性度量,然后构建药物-疾病相似网络,构建药物-疾病异构网络,并在此基础上提出一种双随机游走算法来预测潜在的药物-疾病关联。通过整合药物靶点和疾病基因数据,罗等人。[91]提出了一种新的药物定位方法RWHNDR,用于预测药物与疾病的相互作用。通过整合药物-靶点和疾病-基因的相互作用,他们构建了药物-靶点-疾病网络,并将基本的随机游走模型扩展到所构建的异构网络。
标签传播 考虑到异构网络的标签传播可以有效地探索网络的全局和局部特征,Yan等[92]提出了一种基于网络的标签传播方法LPMIHN来预测DTIs。对于查询药物,LPMIHN首先在药物相似度网络上进行标签传播,然后在具有相互作用信息的目标相似度网络上进行标签传播,以预测所有目标的标签置信度得分。分数超过预设阈值的目标被认为是查询药物的候选对象。
Shahreza等人[93]提出了一种异构标签传播算法Heter-LP,通过整合多源信息来预测潜在的DTIs。首先,将药物相似度、疾病相似度、靶标相似度、DTIs、药物-疾病相互作用和疾病-靶标相互作用相结合,构建一个异构网络。然后,Heter-LP通过异构网络传播标签信息,以识别药物和靶标之间的相互作用。
基于网络的推理 Cheng等[94]基于复杂网络理论,开发了三种监督推理方法,即基于药物的相似推理(drug-based similarity inference, DBSI)、基于目标的相似推理(target-based similarity inference, TBSI)和基于网络的推理(network- Based inference, NBI),用于预测新的药物-靶标关联。其中,NBI可以根据二部药-靶图上的两相扩散对特定药物的候选靶标进行排序,也可以对特定靶标的候选靶标进行排序。NBI通过仅使用已知的药物-目标关联信息,在四个金标准数据集上产生了良好的预测性能。Cheng等[95]通过将加权值分配给边或节点,即边加权的NBI和节点加权的NBI,改进了NBI。系统评价显示,两种加权的NBI方法的表现略优于原始的NBI方法。Alaimo等[96]提出了另一种NBI方法,称为域调谐混合(DT-hybrid),该方法通过相似矩阵添加基于域的知识来扩展NBI和混合算法。将药物相似度、靶标相似度和已知的药物-靶标相关性综合起来,为药物-靶标预测提供了一个统一的框架。计算实验结果表明,DT-hybrid在融合生物知识的DTI预测中明显优于NBI。Wu等[97]开发了一种有用的工具,即亚结构-药物靶标NBI (substruct -drug-target NBI, SDTNBI),用于为旧药物、失败药物和新的化学实体寻找新的靶标。通过整合已知的DTIs、药物-亚结构键和新的化学实体-亚结构键,SDTNBI利用资源扩散方法推断新的DTIs。
Wang等[98]提出了一种基于异质图的推断方法,称为HGBI,用于预测药物-靶标关联。构建包含药物相似度、靶相似度和已知DTIs的药靶异质图。然后,基于关联负罪原理和对异构网络信息流的直观解释,HGBI通过考虑图中连接药物-目标对的所有路径,迭代更新药物-目标对之间的边权值。当更新过程收敛时,得到药物和目标之间的最终权值。Wang等[99]通过同时集成药物、疾病和目标信息,提出了一种新的异构网络模型,该模型将药物-疾病和DTI预测集成到一个统一的计算框架中。Martínez等[100]提出了一种基于网络的优先排序方法——DrugNet,该方法整合了疾病、药物和目标的信息,同时进行药物-疾病和疾病-药物的优先排序。
网络集群和网络路径 Wu等[101]根据已知药物和疾病相关基因及特征信息构建了一个加权的疾病和药物异质性网络,使用两种网络聚类方法寻找包含候选药物-疾病相互作用的模块。Ba-Alawi等人[102]开发了一种计算药物靶标预测方法DASPind,该方法使用从药物靶标异质网络推断出的特定长度的简单路径。基于一种假设,如果有更多的路径连接药物和蛋白质,它们就有更高的可能性相互作用,DASPind跨越了药物和目标蛋白之间的所有简单路径和获得了代表它们之间存在相互作用的可能性的合计分数。
矩阵分解和矩阵补全的方法
为了开发更高效、更准确的算法,来自许多不同领域的各种技术被纳入了生物医学领域[111]。特别是,诸如矩阵分解和矩阵完成等技术已成功地应用于发现新的药物-靶点和药物-疾病相互作用。与其他方法相比,基于推荐模型的计算药物重定位方法不需要负样本,可以灵活地整合更多的先验信息。
基本矩阵分解 矩阵分解方法假设存在决定药物、靶点和疾病关系的有限因素,通过矩阵分解可以有效地得到这些因素。Dai等[103]提出了一种将药物-基因相互作用、疾病-基因相互作用和基因-基因相互作用相结合的矩阵因子分解模型,预测新的药物-疾病关联。考虑到药物和疾病之间的相互作用在基因相互作用网络中有证据,他们将基因组空间整合到所提议的模型中。包括药物-基因相互作用、疾病-基因相互作用和基因-基因相互作用在内的基因组空间可以为鉴定新型药物-疾病关联提供分子生物学信息。Ezzat等[104]提出了两种基于图正则化技术的矩阵分解方法来预测新的DTIs。此外,他们还开发了一种加权k-最近邻方法WKNKN作为预处理步骤,以估计未知药物-目标对的相互作用似然值。
除了药物化学信息和基因序列信息外,还有多种相关信息可以整合,进一步提高药物重新定位的性能。Zheng等[105]利用化学结构、ATC编码、基因组序列、基因本体论和蛋白质-蛋白质相互作用定义了多个药物和靶标相似矩阵,提出了一种多重相似度协同矩阵分解(MSCMF)模型,该模型具有相加正则化项对应相似度信息。MSCMF将药物和目标投影到低秩特征空间中,用于逼近DTIs。Kuang等人[106]提出了一种核矩阵降维(KMDR)方法,利用核矩阵变换预测新的DTIs。KMDR利用化学结构、ATC编码和氨基酸序列信息来定义核的相关药物-靶对。然后,KMDR对定义的核矩阵进行特征值分解,利用前n个最大特征值对应的特征向量构造链接相似矩阵。基于链接相似矩阵和已知的药物-目标对,KMDR识别出DTIs,并优于RLS分类器和半监督链接预测分类器。
贝叶斯矩阵分解 Gönen[107]提出了核化贝叶斯矩阵分解(KBMF2K)方法,它是一种结合降维、矩阵分解和二分类的贝叶斯公式,预测新药和新靶标的相互作用。KBMF2K利用药物化学结构计算药物相似度,将药物相似度矩阵定义为药物核矩阵。 利用蛋白质序列信息计算目标相似度,并将目标相似度矩阵作为目标核矩阵。KBMF2K将药物和靶标投影到一个统一的子空间中,得到药物和靶标的低维特征表示,并以此计算药物-靶标对的相互作用得分。给定一种药物和一个靶标,他们预测的阳性分数表明他们彼此相互作用。KBMF2K可用于预测新药候选靶点、预测新药候选靶点、预测新药与新靶点之间的相互作用。
为了利用对象的多侧信息源,Gönen和Kaski[108]提出了一种新的概率模型KBMF2KMKL,该模型通过融合多核学习扩展了核化矩阵分解。将具有不同权重值的药物(或目标)核线性组合组成联合药物(或目标)核,并将联合药物核和目标核投影到统一的低维空间中。
Logistic矩阵分解 刘等人。[109]提出了一种将Logistic矩阵分解与邻域正则化和邻域平滑相结合的DTI预测算法,即邻域正则化Logistic矩阵分解算法(NRLMF)。在NRLMF中,药物和靶点被投影到一个共享的低维空间,并由潜在向量表示。然后,使用这些潜在向量用Logistic函数对每个药物-靶点对的相互作用概率进行建模。此外,考虑到已知/观察到的药物-靶点对已得到实验验证,NRLMF对已知对的重要性级别高于未知对。以往的基于核的方法往往是将多个核进行线性组合来构造最终的核矩阵。然而,对于那些没有明确线性关系的核函数来说,这是不合理的。Hao等人。[110]提出了一种双网络集成Logistic矩阵分解算法DNILMF来预测新的DTI。对于药物,将化学相似矩阵转化为药物核心矩阵。然后,利用非线性扩散技术将药物核矩阵与推导出的药物高斯核矩阵组合,构成扩散药物矩阵。对于目标,将序列相似度矩阵转换为目标核矩阵,并与推导出的目标高斯核矩阵相结合,构造扩散目标矩阵。然后,DNILMF利用融合相似信息的Logistic矩阵分解来预测药物与目标的相互作用概率。
Lim等人。[111]利用协同过滤技术,提出了一种改进新型化学品目标预测的余弦计算方法。通过应用Logistic矩阵因式分解,余弦将化学物质和蛋白质投影到统一的低维潜在空间来模拟化学-蛋白质相互作用。余弦引入了特定于位置的权重,它本质上考虑了已知或验证的相互作用和相互作用值的推定的重要性。此外,余弦还实现了加权轮廓法来解决冷启动问题。
矩阵补全 矩阵补全方法试图填充药物、目标和疾病关联矩阵中的未知元素,以揭示新的适应症。罗等人。[112]提出了一种药品重新定位推荐系统(DRRS)进行预测潜在的药物适应症,可以有效地解决药物重新定位时的冷启动和稀疏性问题。通过整合包含相似度和交互作用信息的药物和疾病相关数据,构建了药物-疾病矩阵。基于导致药物-疾病相互作用的隐藏因素高度相关的假设,相应的药物-疾病矩阵是低等级的。然后,利用一种快速奇异值阈值算法对未验证相互作用的新的药物-疾病对用预测的相互作用分数来完成药物-疾病矩阵。
Yang等人。[113]提出了一种在低秩假设下完成药物-疾病矩阵的有界核范数正则化(BNNR)方法。虽然BNNR和DRRS是基于相同的药物-疾病矩阵,但BNNR可以通过加入正则化项来处理包含在相似性信息中的噪声数据。此外,BNNR通过加入额外的有界约束,将预测矩阵项值限制在特定区间内,可以得到更多可解释的预测值。他们还开发了一种用于药物重新定位的重叠矩阵补全(OMC)方法[114]。OMC的基本思想是将药物和疾病方面的预测结果结合起来。OMC方法分别为药物-疾病双层网络和药物-蛋白质-疾病三层网络提供了OMC2算法和OMC3算法。缺失的条目使用BNNR模型进行填充。实验结果表明,OMC不仅在交叉验证方面优于其他方法,而且具有更好的计算效率。
此外,Wang et al.。[115]提出了一种拉普拉斯图正则化矩阵补全模型来预测DTI。在他们的工作中,通过使用对偶拉普拉斯图正则化项,药物相似性和靶相似性被纳入完成模型。他们使用基于增广拉格朗日乘子算法的迭代策略解决了矩阵补全问题。
基于深度学习的方法
深度学习是人工神经网络的扩展,它利用多个处理层自动学习具有多层次抽象的数据表示,已成功应用于计算机视觉、语音识别、自然语言处理、化学信息学和生物信息学等领域[116-119]。
考虑到深度学习方法可以提取网络[121]中顶点的拓扑结构特征,提取的特征可以进一步用于计算两个顶点的拓扑相似度。Zong et al.。[120]提出了一种基于深度学习算法DeepWalk[121]的基于相似度的药物靶标预测方法。首先,他们构建了一个以药物、靶标和疾病为顶点,以它们之间的关联为边的异构网络。然后,根据构建的异构网络拓扑结构,利用DeepWalk得到顶点的低维向量表示,并利用它们的表示来计算两个顶点之间的相似度。最后,两种基于相似度的推理方法DBSI和TBSI[94]被用来利用药物-药物相似度和靶-靶相似度来预测新的DTI。
基于药物和靶点的转录组数据,Xie等。[122]将DTI预测建模为二分类任务,并设计了深度神经网络(DNN)模型来预测潜在的相互作用。Wen等人[119]开发了一种基于深度学习的算法框架DeepDTIs,该框架应用深度信念网络来精确预测新的DTIs。药物和靶标的特征分别用分子化学亚结构和蛋白质序列信息来表示。药物靶标对的描述符是通过连接它们的药物和靶标的特征来构建的。然后,将这些描述符作为DeepDTIs的原始输入数据。DeepDTIs通过无监督训练过程从原始输入描述符中学习药物-靶标对的表示,然后从未标记的药物-靶标对中随机抽取阳性药物-靶标对和阴性药物-靶标对建立分类模型。
Öztürk等[123]提出了一种基于深度学习的模型DeepDTA,利用靶标的序列信息和药物的微笑表征来预测DTI的结合亲和力。首先,DeepDTA通过卷积神经网络从微笑和蛋白质序列中学习药物和目标的表示。然后,这些学习到的表示法被用作三个完全连接的层的输入,以预测结合亲和力。
Zeng等[124]通过整合药物相关信息,构建了多个药物网络,利用多模态深度自动编码器学习药物的低维特征。 然后,学习到的特征和与疾病的相互作用被编码和解码,通过一个变种的自动编码器来识别潜在的药物-疾病相互作用。
实验和比较
黄金标准数据集
在药物定位中,两个标准的GOLD数据集主要用于评估预测方法的性能。一个是来自[84]的药物靶标数据集,其中包含一组已知的DTI、药物相似性和蛋白质靶标相似性。在该金标准数据集中,从KEGG BRITE、Brenda、SuperTarget和DrugBank等多个公共数据库收集已知DTI;SIMCOMP根据其化学结构计算药物-药物相似度;靶-靶相似度定义为氨基酸序列相似度,使用归一化版本的Smith-Waterman比对得分计算。化学结构和蛋白质序列数据取自公共数据库KEGG。该金标准数据集根据蛋白质靶标类型分为酶、GPCR、离子通道和核受体四组。每组包含相应的靶点、药物以及药物与靶点的相互作用。
另一个黄金标准数据集是[83]中的药物-疾病数据集,其中包含从DrugBank服用的593种药物与OMIM数据库中列出的313种疾病之间的1933个关联。此外,药物-疾病数据集包含五个药物-药物相似性度量和两个疾病-疾病相似性度量。根据药物化学结构、药物副作用和药物靶点信息实施五种药物-药物相似性度量。基于疾病表型的语义相似度来度量成对的疾病-疾病相似度。在随后的评价实验中,该金标准药物-疾病数据集与一些研究中使用的F数据集相对应。此外,在[90,100]中还收集了另外两个药病数据集,包括CDATA集和DNdata集。CDATA集包含663种药物、409种疾病和2352对相互作用的药物-疾病对。DNA数据集包含1490种药物、4516种疾病和1008对相互作用的药物-疾病对。
评估指标
对于前面提到的大多数计算方法,预测方法通常是基于n次交叉验证或遗漏一次交叉验证来评估的。N重CV实验通常以两种不同的方式进行:(1)交互预测(CV-Pair):将所有已知或已验证的交互随机分成n个折叠。每轮以(n−1)/n个相互作用作为训练集,剩余的1/n个相互作用作为测试集;(2)新药预测(CV-Druits):将所有药物分为n个文件夹。每轮使用(n−1)/n的药物作为训练集,剩余的1/n的药物作为测试集。对于遗漏的CV,通过选择一个交互作用作为测试集进行交互预测,将剩余的交互作用作为训练集;通过选择一个药物作为测试集进行新药预测,将剩余的药物作为训练集。没有已知相互作用的药物-靶点或药物-疾病对被认为是候选对。
受试者工作特征(ROC)曲线绘制了不同阈值下的真阳性率与假阳性率之间的关系。准确率-召回率(PR)曲线绘制了不同阈值下的准确率-召回率之间的关系。在交叉验证实验中,药物重新定位方法预测所有候选药物-靶点或药物-疾病对的相互作用概率。真阳性是正确预测的已知交互对的数量,假阳性是错误预测的非交互对的数量。在进行交叉验证后,计算ROC曲线下面积(AUC)和PR曲线下面积(AUPR)来评价不同方法的性能。对于两种常用的评估指标,据报道,对于高度不平衡的数据集,AUPR可以提供比AUC更多的信息评估[125]。
实验结果
通过交叉验证实验,比较了各种预测方法在药物靶点和药物疾病金标准数据集上的性能。对于药物靶标预测任务,从相应研究[92,109,110,126]获得的AUC和AuPr值的实验结果显示在表4和5中,其中每行中的最佳实验结果用粗体表示,次佳结果用下划线表示。从表4可以看出,NRLMF具有更好的预测性能。NRLMF的成功可以归功于邻域正则化Logistic分解的使用,它对已知/阳性的DTI给予了更高的重视,并通过邻域正则化考虑了药物和靶点最近邻域的影响。DNILMF采用非线性扩散技术构造扩散药物相似度矩阵和目标相似度矩阵。然后,DNILMF结合扩散相似度和DTI信息对交互概率得分进行预测,取得了最好的预测结果。
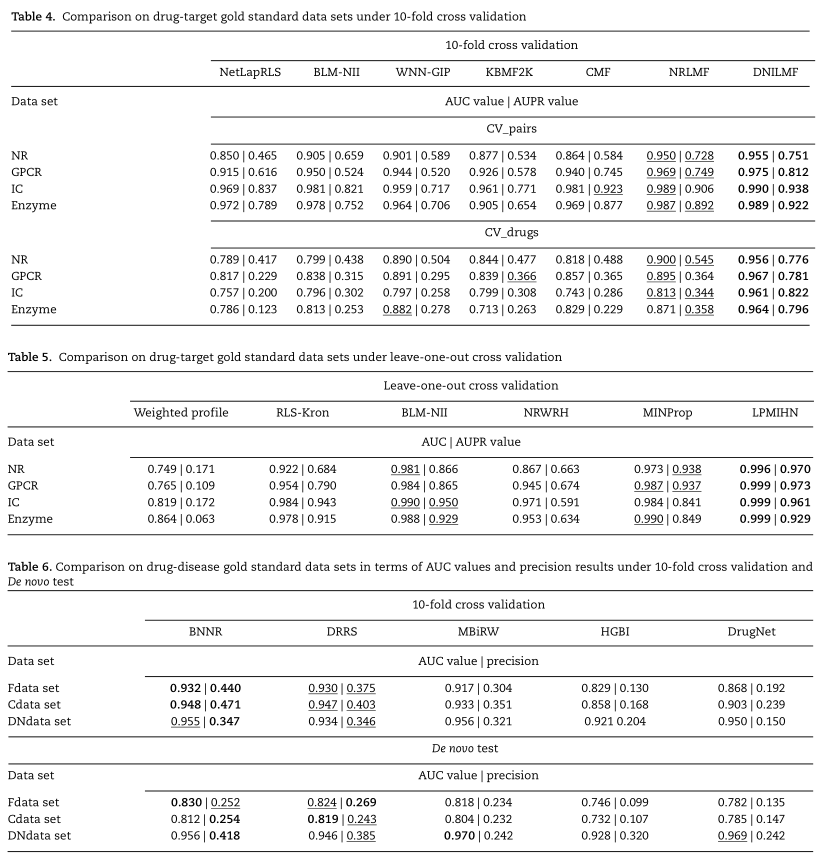
此外,在表5中可以观察到,具有来自异构网络的相互作用信息的标签传播(LPMIHN)倾向于主导其他标签传播,并且MINProp获得次优结果。实验结果表明,LPMIHN方法将相似度信息与交互关系的拓扑信息相结合的策略有助于提高预测性能。此外,MINProp也是一种基于标签传播的预测方法,与LPMIHN在标签传播路径上有所不同。
对于药物-疾病预测任务,以AUC和精度表示的实验结果是从我们先前的研究[113]中获得的,如表6所示。[113]中的从头检验是通过选择只有一种已知相互作用的药物来评估预测方法识别没有验证相互作用的新药的新适应症的能力。人们可以发现,基于矩阵补全模型设计的BNNR和DRRS在10倍CV和从头检验中都比其他方法在所有数据集上都取得了更好的预测性能。这也表明BNNR和DRRS为冷启动问题提供了有效的解决方案,冷启动问题是新药在药物重新定位中的预测。
挑战与未来工作
药物重新定位是药物发现和开发的一种重要而有前途的方法学。与传统的药物发现相比,药物重新定位可以缩短时间,节约成本,降低失败的概率,因为它从具有众所周知的安全性和药动学特征的候选化合物开始[4,127]。在大规模生物数据和各种模型的基础上,实现了包括药物-靶点识别和药物-疾病相互作用识别在内的计算药物定位。本文对药物重新定位涉及的生物和医学数据、公共数据库、计算预测方法、评价指标和常用的金标准数据集进行了总结和讨论。
药物定位是一个复杂的过程,生物信息的准确性和计算模型的准确性会影响预测方法的性能。我们从数据的角度回顾了与药物重新定位相关的公共数据库和最近的应用。可以发现,在最近的研究中已经利用了各种类型的生物医学数据,如化学结构、生物活性图谱、副作用、治疗效果、基因表达、药物结合位点、药物相互作用、本体和语义数据。然而,这些生物医学数据在实际应用中往往是不确定的,因为数据噪声高、不完整、不准确。通过充分利用来自不同异构数据源的各种生物和医学数据,计算预测模型可以实现对药物-靶点和药物-疾病相互作用的更准确的识别。通常,一种类型的数据仅反映生物医学实体的一个或几个方面。未来的努力应该更彻底地将海量和异构的可用数据(化学、生物、结构、临床)集成到一个统一的工作流程中,这显然是一项具有挑战性的任务。对于数据集成,药物定位方法应考虑不同生物医学数据的不同贡献,并设计合理的权重分配机制进行改进。
通过利用这些数据,基于各种计算模型的许多预测方法已被应用于识别药物-靶点或药物-疾病相互作用。我们根据这些预测方法所采用的计算模型对它们进行了分组。根据最近报道的研究,在这些方法中,矩阵分解和基于矩阵补全的方法表现出了较好的预测性能,多源信息的集成可以进一步提高计算药物定位方法的预测精度。一般来说,每种计算方法都有其优点、适用性、缺点和局限性。例如,一些方法可以预测具有已知相互作用的已批准药物的候选药物,但不能预测没有任何已知相互作用的新药的候选药物,并且一些方法不能集成多种类型的生物药物重新定位中的信息。因此,通过分析它们的特点,可以将各种计算方法有效地组合在一起。从而进一步扩大了它们的适用范围,提高了它们的预测精度。
在实际应用中,已验证或已标记的药物-靶点和药物-疾病相互作用的数量远远少于未标记的相互作用,这就带来了药物重新定位中的数据稀疏问题。因此,可以利用生物医学实体的一些先验信息对交互数据进行预处理,并补充缺失值,以缓解稀疏性问题。一些药物定位方法需要正负样本来训练预测模型。然而,在实际应用中,负样本往往是稀缺或缺失的,大多数研究都是通过从未标注的数据中随机抽取负样本来解决这一问题,这可能会影响预测模型的性能。因此,应该设计合理的负样本选择方法来生成训练集中使用的RN样本。为了公平地比较和评价各种计算药物定位模型,需要从多个数据源中收集有用信息,构建包含全面生物医学数据的基准数据集。分析和解释预测的新相互作用的潜在机制是药物重新定位中的一个重要问题。许多研究通过从现有的公共数据库和文献中搜索证据来进行案例研究,以分析预测的相互作用。但是这样的分析,需要专业知识,而且非常耗时。因此,需要更多的研究来解决药物定位方法预测的新的相互作用的验证问题。
关键点 **· **多种类型的生物医学数据可以被利用和整合,以开发计算药物重新定位方法,并验证其预测结果。本文从机器学习的角度对现有的潜在药物-靶点或药物-疾病相互作用预测的计算方法进行了分析和分类。 **·**通过从多个数据源收集生物和医学信息,可以构建全面的基准数据集。 **·**合理的负样本选择方法可以产生可靠的负样本来训练预测模型。
References
- Li J, Zheng S, Chen B, et al. A survey of current trends in com-putational drug repositioning. Brief Bioinform 2015;17:2–12.
- Hurle M, Yang L, Xie Q, et al. Computational drug repo-sitioning: from data to therapeutics. Clin Pharmacol Ther2013;93:335–41.
- Kim T-W. Drug repositioning approaches for the discovery of new therapeutics for Alzheimer’s disease. Neurotherapeu- tics 2015;12:132–42.
- Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov 2004;3:673.
- Vanhaelen Q, Mamoshina P, Aliper AM, et al. Design of efficient computational workflows for in silico drug repur- posing. Drug Discov Today 2017;22:210–22.
- Hernandez JJ, Pryszlak M, Smith L, et al. Giving drugs a second chance: overcoming regulatory and financial hur- dles in repurposing approved drugs as cancer therapeutics. Front Oncol 2017;7:273.
- Shim JS, Liu JO. Recent advances in drug repositioning for the discovery of new anticancer drugs. Int J Biol Sci 2014;10:654.
- Turanli B, Grøtli M, Boren J, et al. Drug repositioning for effective prostate cancer treatment. Front Physiol 2018;9.
- Zou J, Zheng M-W, Li G, et al. Advanced systems biology methods in drug discovery and translational biomedicine. Biomed Res Int 2013;2013:742835.
- Lavecchia A, Cerchia C. In silico methods to address polypharmacology: current status, applications and future perspectives. Drug Discov Today 2016;21:288–98.
- Keiser MJ, Roth BL, Armbruster BN, et al. Relating pro- tein pharmacology by ligand chemistry. Nat Biotechnol 2007;25:197.
- Acharya C, Coop A, E Polli J, et al. Recent advances in ligand- based drug design: relevance and utility of the conforma- tionally sampled pharmacophore approach. Curr Comput Aided Drug Des 2011;7:10–22.
- Cheng AC, Coleman RG, Smyth KT, et al. Structure-based maximal affinity model predicts small-molecule drugga- bility. Nat Biotechnol 2007;25:71.
- Napolitano F, Zhao Y, Moreira VM, et al. Drug repositioning: a machine-learning approach through data integration. J Chem 2013;5:30.
- Li J, Lu Z. A new method for computational drug reposition- ing using drug pairwise similarity. In: 2012 IEEE International Conference on Bioinformatics and Biomedicine. IEEE, 2012, 1–4.
- March-Vila E, Pinzi L, Sturm N, et al. On the integration of in silico drug design methods for drug repurposing. Front Pharmacol 2017;8:298.
- Yella J, Yaddanapudi S, Wang Y, et al. Changing trends in computational drug repositioning. Pharm 2018;11:57.
- Ding H, Takigawa I, Mamitsuka H, et al. Similarity-based machine learning methods for predicting drug–target inter- actions: a brief review. Brief Bioinform 2013;15:734–47.
- Zhu Y, Elemento O, Pathak J, et al. Drug knowledge bases and their applications in biomedical informatics research. Brief Bioinform 2019;20:1308–21.
- Chen X, Yan CC, Zhang X, et al. Drug–target interaction pre- diction: databases, web servers and computational models. Brief Bioinform 2016;17:696–712.
- Hao M, Bryant SH, Wang Y. Open-source chemogenomic data-driven algorithms for predicting drug–target interac- tions. Brief Bioinform 2019;20:1465–74.
- Ezzat A, Wu M, Li X-L, et al. Computational prediction of drug-target interactions using chemogenomic approaches: an empirical survey. Brief Bioinform 2019;20:1337–57.
- Lotfi Shahreza M, Ghadiri N, Mousavi SR, et al. A review of network-based approaches to drug repositioning. Brief Bioinform 2017;19:878–92.
- Rifaioglu AS, Atas H, Martin MJ, et al. Recent applications of deep learning and machine intelligence on in silico drug discovery: methods, tools and databases. Brief Bioinform 2019;20:1878–912.
- Zhou L, Li Z, Yang J, et al. Revealing drug-target interac- tions with computational models and algorithms. Molecules 2019;24:1714.
- Liu T, Lin Y, Wen X, et al. BindingDB: a web-accessible database of experimentally determined protein–ligand binding affinities. Nucleic Acids Res 2006;35:D198–201.
- Gaulton A, Bellis LJ, Bento AP, et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res 2011;40:D1100–7.
- Law V, Knox C, Djoumbou Y, et al. DrugBank 4.0: shed- ding new light on drug metabolism. Nucleic Acids Res 2013;42:D1091–7.
- Fu C, Jin G, Gao J, et al. DrugMap Central: an on-line query and visualization tool to facilitate drug repositioning stud- ies. Bioinformatics 2013;29:1834–6. Downloaded from https://academic.oup.com/bib/advance-article/doi/10.1093/bib/bbz176/5732424 by Henan University user on 29 January 2021 14 Luo et al.
- Drugs.com. http://www.drugs.com/.
- Tatonetti NP, Patrick PY, Daneshjou R, et al. Data-driven prediction of drug effects and interactions. Sci Transl Med 2012;4:125ra131–1.
- Von Eichborn J, Murgueitio MS, Dunkel M, et al. PROMIS- CUOUS: a database for network-based drug-repositioning. Nucleic Acids Res 2010;39:D1060–6.
- Kim S, Thiessen PA, Bolton EE, et al. PubChem substance and compound databases. Nucleic Acids Res 2015;44:D1202–
- Kuhn M, Letunic I, Jensen LJ, et al. The SIDER database of drugs and side effects. Nucleic Acids Res 2015;44:D1075–9.
- Kuhn M, von Mering C, Campillos M, et al. STITCH: interac- tion networks of chemicals and proteins. Nucleic Acids Res 2007;36:D684–8.
- Günther S, Kuhn M, Dunkel M, et al. SuperTarget and Matador: resources for exploring drug-target relationships. Nucleic Acids Res 2007;36:D919–22.
- Kibbe WA, Arze C, Felix V, et al. Disease ontology 2015 update: an expanded and updated database of human diseases for linking biomedical knowledge through disease data. Nucleic Acids Res 2014;43:D1071–8.
- Pletscher-Frankild S, Pallejà A, Tsafou K, et al. DISEASES: text mining and data integration of disease–gene associ- ations. Methods 2015;74:83–9.
- Liu C-C, Tseng Y-T, Li W, et al. DiseaseConnect: a compre- hensive web server for mechanism-based disease–disease connections. Nucleic Acids Res 2014;42:W137–46.
- Piñero J, Bravo À, Queralt-Rosinach N, et al. DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res 2017;45:D833–D839.
- Babbi G, Martelli PL, Profiti G, et al. eDGAR: a database of Disease-Gene Associations with annotated Relationships among genes. BMC Genomics 2017;18:554.
- Becker KG, Barnes KC, Bright TJ, et al. The genetic associa- tion database. Nat Genet 2004;36:431.
- Köhler S, Vasilevsky NA, Engelstad M, et al. The human phenotype ontology in 2017. Nucleic Acids Res 2016;45:D865–
- World Health Organization. ICD-11 for Mortality and Mor- bidity Statistics (ICD-11 MMS) 2018 version. Available at: https://icd.who.int/browse11/l-m/en.
- Rappaport N, Twik M, Plaschkes I, et al. MalaCards: an amal- gamated human disease compendium with diverse clinical and genetic annotation and structured search. Nucleic Acids Res 2016;45:D877–87.
- Lipscomb CE. Medical subject headings (MeSH). Bull Med Libr Assoc 2000;88:265.
- Hamosh A, Scott AF, Amberger JS, et al. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res 2005;33: D514–7.
- Bodenreider O. The unified medical language system (UMLS): integrating biomedical terminology. Nucleic Acids Res 2004;32:D267–70.
- Stark C, Breitkreutz B-J, Reguly T, et al. BioGRID: a gen- eral repository for interaction datasets. Nucleic Acids Res 2006;34:D535–9.
- Consortium GO. The gene ontology (GO) database and informatics resource. Nucleic Acids Res 2004;32:D258–61.
- Keshava Prasad T, Goel R, Kandasamy K, et al. Human protein reference database—2009 update. Nucleic Acids Res 2008;37:D767–72.
- Rose PW, Prli´c A, Altunkaya A, et al. The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic Acids Res 2016;gkw1000.
- Mering C, Huynen M, Jaeggi D, et al. STRING: a database of predicted functional associations between proteins. Nucleic Acids Res 2003;31:258–61.
- Apweiler R, Bairoch A, Wu CH, et al. UniProt: the universal protein knowledgebase. Nucleic Acids Res 2004;32:D115–9.
- Davis AP, Grondin CJ, Johnson RJ, et al. The comparative toxicogenomics database: update 2017. Nucleic Acids Res 2016;45:D972–8.
- Kanehisa M, Furumichi M, Tanabe M, et al. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 2016;45:D353–61.
- Thorn CF, Klein TE, Altman RB. PharmGKB: the phar- macogenomics knowledge base. Methods Mol Biol 2013; 311–20.
- Deng Z, Tu W, Deng Z, et al. PhID: An open-access integrated pharmacology interactions database for drugs, targets, dis- eases, genes, side-effects, and pathways. J Chem Inf Model 2017;57:2395–400.
- Li YH, Yu CY, Li XX, et al. Therapeutic target database update 2018: enriched resource for facilitating bench-to- clinic research of targeted therapeutics. Nucleic Acids Res 2017;46:D1121–7.
- Nicola G, Liu T, Gilson MK. Public domain databases for medicinal chemistry. J Med Chem 2012;55:6987–7002.
- Steinbeck C, Han Y, Kuhn S, et al. The Chemistry Development Kit (CDK): An open-source Java library for chemo-and bioinformatics. J Chem Inf Comput Sci 2003;43: 493–500.
- Hattori M, Tanaka N, Kanehisa M, et al. SIMCOMP/SUB- COMP: chemical structure search servers for network anal- yses. Nucleic Acids Res 2010;38:W652–6.
- Perlman L, Gottlieb A, Atias N, et al. Combining drug and gene similarity measures for drug-target elucidation. J Com- put Biol 2011;18:133–45.
- Hodos RA, Kidd BA, Khader S, et al. Computational approaches to drug repurposing and pharmacology. Wiley Interdiscip Rev Syst Biol Med 2016;8:186.
- Vilar S, Uriarte E, Santana L, et al. Similarity-based model- ing in large-scale prediction of drug-drug interactions. Nat Protoc 2014;9:2147.
- Zhou L, Plasek JM, Mahoney LM, et al. Mapping partners master drug dictionary to RxNorm using an NLP-based approach. J Biomed Inform 2012;45:626–33.
- Rodriguez-Esteban R. A drug-centric view of drug devel- opment: how drugs spread from disease to disease. PLoS Comput Biol 2016;12:e1004852.
- Wang Y, Chen S, Deng N, et al. Drug repositioning by kernel- based integration of molecular structure, molecular activ- ity, and phenotype data. PLoS One 2013;8:e78518.
- Schuffenhauer A, Floersheim P, Acklin P, et al. Similarity metrics for ligands reflecting the similarity of the target proteins. J Chem Inf Comput Sci 2003;43:391–405.
- Chiang AP, Butte AJ. Systematic evaluation of drug–disease relationships to identify leads for novel drug uses. Clinical Pharmacology & Therapeutics 2009;86:507–10.
- Bleakley K, Yamanishi Y. Supervised prediction of drug– target interactions using bipartite local models. Bioinformat- ics 2009;25:2397–403.
- Wang Y-C, Zhang C-H, Deng N-Y, et al. Kernel-based data fusion improves the drug–protein interaction prediction. Comput Biol Chem 2011;35:353–62. Downloaded from https://academic.oup.com/bib/advance-article/doi/10.1093/bib/bbz176/5732424 by Henan University user on 29 January 2021 Biomedical data and computational models for drug repositioning 15
- Liu H, Sun J, Guan J, et al. Improving compound–protein interaction prediction by building up highly credible neg- ative samples. Bioinformatics 2015;31:i221–9.
- Liu H, Song Y, Guan J, et al. Inferring new indications for approved drugs via random walk on drug-disease hetero- geneous networks. BMC Bioinformatics 2016;17:539.
- Lan W, Wang J, Li M, et al. Predicting drug–target inter- action using positive-unlabeled learning. Neurocomputing 2016;206:50–7.
- Peng L, Zhu W, Liao B, et al. Screening drug-target interac- tions with positive-unlabeled learning. Sci Rep 2017;7:8087.
- Xia Z, Wu L-Y, Zhou X, et al. Semi-supervised drug- protein interaction prediction from heterogeneous biologi- cal spaces. In: BMC Systems Biology BioMed Central, 2010, S6.
- van Laarhoven T, Nabuurs SB, Marchiori E. Gaussian inter- action profile kernels for predicting drug–target interac- tion. Bioinformatics 2011;27:3036–43.
- Nascimento AC, Prudêncio RB, Costa IG. A multiple kernel learning algorithm for drug-target interaction prediction. BMC bioinformatics 2016;17:46.
- Van Laarhoven T, Marchiori E. Predicting drug-target inter- actions for new drug compounds using a weighted nearest neighbor profile. PLoS One 2013;8:e66952.
- Hao M, Wang Y, Bryant SH. Improved prediction of drug- target interactions using regularized least squares inte- grating with kernel fusion technique. Anal Chim Acta 2016;909:41–50.
- Chen H, Li J. A flexible and robust multi-source learning algorithm for drug repositioning. In: Proceedings of the 8th ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics, ACM, 2017, 510–5.
- Gottlieb A, Stein GY, Ruppin E, et al. PREDICT: a method for inferring novel drug indications with application to personalized medicine. Mol Syst Biol 2011;7.
- Yamanishi Y, Araki M, Gutteridge A, et al. Prediction of drug–target interaction networks from the integration of chemical and genomic spaces. Bioinformatics 2008;24: i232–40.
- Yamanishi Y, Kotera M, Kanehisa M, et al. Drug-target interaction prediction from chemical, genomic and phar- macological data in an integrated framework. Bioinformatics 2010;26:i246–54.
- Olayan RS, Ashoor H, Bajic VB. DDR: efficient computa- tional method to predict drug–target interactions using graph mining and machine learning approaches. Bioinfor- matics 2017;34:1164–73.
- Cao DS, Zhang LX, Tan GS, et al. Computational prediction of drug-target interactions using chemical, biological, and network features. Mol Inform 2014;33:669–81.
- Huang Y-F, Yeh H-Y, Soo V-W. Inferring drug-disease asso- ciations from integration of chemical, genomic and phe- notype data using network propagation. BMC Med Genet 2013;6:S4.
- Chen X, Liu M-X, Yan G-Y. Drug–target interaction predic- tion by random walk on the heterogeneous network. Mol BioSyst 2012;8:1970–8.
- Luo H, Wang J, Li M, et al. Drug repositioning based on comprehensive similarity measures and Bi-Random walk algorithm. Bioinformatics 2016;32:2664–71.
- Luo H, Wang J, Li M, et al. Computational drug repositioning with random walk on a heterogeneous network. IEEE/ACM Trans Comput Biol Bioinform.
- Yan X-Y, Zhang S-W, Zhang S-Y. Prediction of drug–target interaction by label propagation with mutual interaction information derived from heterogeneous network. Mol BioSyst 2016;12:520–31.
- Shahreza ML, Ghadiri N, Mousavi SR, et al. Heter-LP: a heterogeneous label propagation algorithm and its appli- cation in drug repositioning. J Biomed Inform 2017;68: 167–83.
- Cheng F, Liu C, Jiang J, et al. Prediction of drug-target inter- actions and drug repositioning via network-based infer- ence. PLoS Comput Biol 2012;8:e1002503.
- Cheng F, Zhou Y, Li W, et al. Prediction of chemical-protein interactions network with weighted network-based infer- ence method. PLoS One 2012;7:e41064.
- Alaimo S, Pulvirenti A, Giugno R, et al. Drug–target inter- action prediction through domain-tuned network-based inference. Bioinformatics 2013;29:2004–8.
- Wu Z, Cheng F, Li J, et al. SDTNBI: an integrated network and chemoinformatics tool for systematic prediction of drug– target interactions and drug repositioning. Brief Bioinform 2016;18:333–47.
- Wang W, Yang S, Li J. Drug target predictions based on heteroge- neous graph inference. Biocomputing 2013 World Scientific, 2013, 53–64.
- Wang W, Yang S, Zhang X, et al. Drug repositioning by integrating target information through a heterogeneous network model. Bioinformatics 2014;30:2923–30.
- Martínez V, Navarro C, Cano C, et al. DrugNet: network- based drug–disease prioritization by integrating heteroge- neous data. Artif Intell Med 2015;63:41–9.
- Wu C, Gudivada RC, Aronow BJ, et al. Computational drug repositioning through heterogeneous network clustering. BMC Syst Biol 2013;7:S6.
- Ba-Alawi W, Soufan O, Essack M, et al. DASPfind: new efficient method to predict drug–target interactions. J Chem 2016;8:15.
- Dai W, Liu X, Gao Y, et al. Matrix factorization-based predic- tion of novel drug indications by integrating genomic space. Comput Math Methods Med 2015;2015:275045.
- Ezzat A, Zhao P, Wu M, et al. Drug-target interaction predic- tion with graph regularized matrix factorization. IEEE ACM T Comput Bi 2017;14:646–56.
- Zheng X, Ding H, Mamitsuka H, et al. Collaborative matrix factorization with multiple similarities for predicting drug- target interactions. In: Proceedings of the 19th ACM SIGKDD international conference on Knowledge discovery and data min- ing. ACM, 2013, 1025–33.
- Kuang Q, Li Y, Wu Y, et al. A kernel matrix dimension reduction method for predicting drug-target interaction. Chemom Intell Lab Syst 2017;162:104–10.
- Gönen M. Predicting drug–target interactions from chemi- cal and genomic kernels using Bayesian matrix factoriza- tion. Bioinformatics 2012;28:2304–10.
- Gönen M, Khan S, Kaski S. Kernelized Bayesian matrix factorization. In: International Conference on Machine Learning, 2013, 864–72.
- Liu Y, Wu M, Miao C, et al. Neighborhood regularized logistic matrix factorization for drug-target interaction prediction. PLoS Comput Biol 2016;12:e1004760.
- Hao M, Bryant SH, Wang Y. Predicting drug-target interac- tions by dual-network integrated logistic matrix factoriza- tion. Sci Rep 2017;7:40376.
- Lim H, Gray P, Xie L, et al. Improved genome-scale multi-target virtual screening via a novel collaborative filtering approach to cold-start problem. Sci Rep 2016;6:
Downloaded from https://academic.oup.com/bib/advance-article/doi/10.1093/bib/bbz176/5732424 by Henan University user on 29 January 2021 16 Luo et al. 112. Luo H, Li M, Wang S, et al. Computational drug reposition- ing using low-rank matrix approximation and randomized algorithms. Bioinformatics 2018;34:1904–12. 113. Yang M, Luo H, Li Y, et al. Drug repositioning based on bounded nuclear norm regularization. Bioinformatics 2019;35:i455–63. 114. Yang M, Luo H, Li Y, et al. Overlapped matrix completion for predicting drug-associated indications. PLoS Comput Biol 2019; doi:10.1371/journal.pcbi.1007541. 115. Wang M, Tang C, Chen J. Drug-target interaction prediction via dual laplacian graph regularized matrix completion. Biomed Res Int 2018;2018:1425608. 116. LeCun Y, Bengio Y, Hinton G. Deep learning. Nature 2015;521:436. 117. Singaravel S, Suykens J, Geyer P. Deep-learning neural- network architectures and methods: Using component- based models in building-design energy prediction. Adv Eng Inform 2018;38:81–90. 118. Xu Y, Dai Z, Chen F, et al. Deep learning for drug-induced liver injury. J Chem Inf Model 2015;55:2085–93. 119. Wen M, Zhang Z, Niu S, et al. Deep-learning-based drug– target interaction prediction. J Proteome Res 2017;16:1401–9. 120. Zong N, Kim H, Ngo V, et al. Deep mining heterogeneous networks of biomedical linked data to predict novel drug– target associations. Bioinformatics 2017;33:2337–44. 121. Perozzi B, Al-Rfou R. Skiena S. Deepwalk: Online learning of social representations. In: Proceedings of the 20th ACM SIGKDD international conference on Knowledge discovery and data mining. ACM, 2014, 701–10. 122. Xie L, Zhang Z, He S, et al. Drug-target interaction prediction with a deep-learning-based model. In: 2017 IEEE Interna- tional Conference on Bioinformatics and Biomedicine (BIBM). IEEE, 2017, 469–76. 123. Öztürk H, Özgür A, Ozkirimli E. DeepDTA: deep drug– target binding affinity prediction. Bioinformatics 2018;34: i821–9. 124. Zeng X, Zhu S, Liu X, et al. deepDR: a network-based deep learning approach to in silico drug repositioning. Bioinfor- matics 2019; doi:10.1093/bioinformatics/btz418. 125. Saito T, Rehmsmeier M. The precision-recall plot is more informative than the ROC plot when evaluating binary classifiers on imbalanced datasets. PLoS One 2015;10: e0118432. 126. Mei J-P, Kwoh C-K, Yang P, et al. Drug–target interaction pre- diction by learning from local information and neighbors. Bioinformatics 2012;29:238–45. 127. Nagaraj A, Wang Q, Joseph P, et al. Using a novel computa- tional drug-repositioning approach (DrugPredict) to rapidly identify potent drug candidates for cancer treatment. Onco- gene 2018;37:403.