9.7 KiB
| title | date | updated | tags | categories | keywords | description | top_img | comments | cover | toc | toc_number | toc_style_simple | copyright | copyright_author | copyright_author_href | copyright_url | copyright_info | katex | highlight_shrink | aside |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Heterogeneous graph inference with matrix completion for computational drug repositioning | 2020-12-29 10:04:11 | 2021-12-19 13:55:41 | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | <nil> | true | <nil> | <nil> |
Abstract
动机:新的证据表明传统的药物发现实验耗时且成本高。计算药物重新定位在为药物研究和发现节省时间和资源方面起着至关重要的作用。因此,制定更准确、更有效的方法势在必行。异构图推理是计算药物重新定位的经典方法,它不仅具有较高的收敛精度,而且具有较快的收敛速度。然而,该方法没有充分考虑异构关联网络的稀疏性。此外,粗略的相似性度量可以降低识别药物相关适应症的性能。 结果:在这篇文章中,我们提出了一种基于矩阵完成的异质图推理(HGIMC)方法来预测批准药物和新药的潜在适应症。首先,我们使用有界矩阵完成(BMC)模型来预填充原始药物-疾病关联矩阵中的一部分缺失条目。这一步可以在药物网络和疾病网络之间添加更多积极的和形成性的药物-疾病边缘。其次,由于异构图推理的性能更依赖于相似性度量,因此采用高斯径向基函数(GRB)来改善药物和疾病的相似性。接下来,基于最新的药物-疾病关联和新的药物与疾病相似性度量,我们构建了一个新的异质药物-疾病网络。最后,HGIMC利用异构网络推断未知关联对的分数,然后推荐有前景的药物适应症。为了评估我们方法的性能,在10倍交叉验证和从头试验中,将HGIMC与五种最先进的药物重新定位方法进行比较。数值结果表明,HGIMC不仅具有较好的预测性能,而且具有较高的计算效率。此外,案例研究也证实了该方法在实际应用中的有效性。 可用性和实施:HGIMC软件和数据可在https://github.com/BioinformaticsCSU/HGIMC, https://hub.docker.com/repository/docker/yangmy84/hgimc and http://doi.org/10.5281/zenodo.4285640. 联系人: jxwang@mail.csu.edu.cn Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
传统的药物发现耗时、复杂且昂贵(Chong等人,2007;Pushpakom等人,2019年)。在过去几十年中,药物研发的投资大幅增加,但市场上的新药数量仍然很低。事实上,I期临床试验中约90%的候选药物失败(Li等人,2016年)。药物重新定位被认为是一种很有前途的药物开发策略,其目标是为现有药物寻找新的候选适应症。由于这些已知药物已经通过了各种临床试验并确保了安全性,因此可以跳过重新定位药物的第一阶段临床试验。因此,药物重新定位可以缩短药物研究周期,降低成本。从数据库(Tanoli等人,2020年)、方法(Luo等人,2020年)和趋势(Karaman和Sippl,2019年)的角度来看,最近对药物重新定位进行了一些综述。
到目前为止,已经提出了许多计算药物重新定位方法,大致可分为三类,包括基于机器学习的方法、基于网络传播的方法和基于推荐系统的方法。这些方法基于一个假设,即相似的药物与相似的疾病相关,反之亦然。在基于机器学习的方法中,潜在药物-疾病关联的预测可以被视为一个二元分类问题。药物-疾病关联被视为样本,而药物和疾病的先前相似性被视为特征。受临床副作用提供药物的人类表型特征这一事实的推动,Yang和Agarwal(2011)扩展了Naive Bayes模型,以确定使用副作用作为特征的临床药物的潜在适应症。综合药物和疾病的多重相似性,Gottlieb等人(2011年)使用逻辑回归模型预测有希望的药物-疾病关联。此外,还使用支持向量机(SVM)分类器验证已知药物的治疗类别(Napolitano等人,2013)。深度学习是机器学习的一个新的分支领域,近年来被用于计算药物重新定位。Aliper等人(2016年)使用完全连接的深层神经网络识别药物的潜在新适应症。该模型在分类精度上优于支持向量机。
基于网络传播的方法可以推断出异构网络中的缺失边,在计算效率上有很大的优势。根据关联犯罪原则,王某等人提出了自己的观点。(2013)提出了一种基于异构图的推理(HGBI)算法,用于预测与药物相关的目标。该算法利用异构图中的所有路径更新交互对之间的边权重,不仅具有较高的收敛精度,而且具有较快的收敛速度。HGBI算法也被扩展以识别新的候选药物适应症(Wang等人,2014年b)。Martinez等人。(2015)提出了一种基于网络的优先排序方法,称为DrugNet,用于预测现有药物的新疾病。通过构建一个异构的药物-目标-疾病网络,DrugNet能够利用传播流实现药物-疾病优先顺序和疾病-药物优先顺序。 通过利用稀疏的药物-疾病关联来提高药物和疾病的相似性度量,罗等人。(2016)开发了一种双随机行走算法,即MBiRW,用于执行关联预测。
对于基于推荐系统的方法,预测药物与疾病关联的问题被认为是一个用户-项目评分问题。矩阵补全和矩阵分解是推荐系统中最常用的两种方法。Luo等人。(2018)提出了一个药物重新定位推荐系统(DRRS),以预测已知和新药的有前途的适应症。DRRS使用奇异值阈值(SVT)算法(Cai等人,2010)完成了由药物相似矩阵、疾病相似矩阵和药物-疾病关联矩阵组成的全局邻接矩阵。基于DRRS的邻接矩阵,Yang et al.。 (2019a)提出了一种低秩矩阵补全方法,即有界核范数正则化方法(BNNR)。BNNR可以通过整合正则项来处理药物和疾病相似性带来的噪声。此外,有界约束可以确保所有完成的值都在特定的间隔内。为了整合多种类型的药物和疾病信息,杨等人。(2019b)提出了用于三层异构网络(OMC3)的重叠矩阵补全。Xuan等人。(2019)提出了一种非负矩阵分解方法,称为DisDrugPred。为了充分利用有用的潜在信息,DisDrugPred考虑了药物和疾病的多重相似性以及药物-疾病对的稀疏性。
受BNNR的低秩完成和HGBI的负罪关联原理的启发,我们提出了一种基于矩阵完成的异质图推理(HGIMC)方法来预测新药相关适应症。HGIMC的主要思想是克服HGBI模型的两个缺陷,包括极度稀疏的异构关联网络和不太精确的相似矩阵。具体来说,首先,我们使用现有的软件包计算药物相似性的五个度量和疾病相似性的两个度量,并获得它们的平均值。然后,我们使用有界矩阵完成(BMC)以高置信度预填充一些药物-疾病关联,以丰富药物网络和疾病网络之间的边缘。此外,我们将高斯径向基函数(GRB)应用于平均相似性矩阵,以获得更好的相似性度量。最后,结合这两个综合相似矩阵和更新后的药物-疾病关联矩阵,异质图推理可以快速准确地预测潜在的药物-疾病关联。HGIMC模型的总体流程图如图1所示。
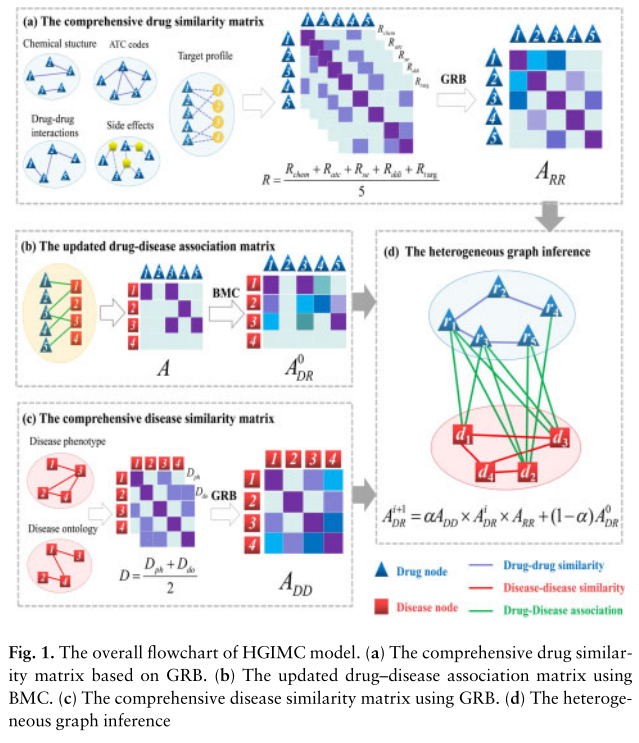 图1.HGIMC模型总体流程图。(A)基于GRB的综合药物相似度矩阵。(B)使用BMC更新的药物-疾病关联矩阵。(C)基于GRB的综合疾病相似度矩阵。(D)异构图推理
图1.HGIMC模型总体流程图。(A)基于GRB的综合药物相似度矩阵。(B)使用BMC更新的药物-疾病关联矩阵。(C)基于GRB的综合疾病相似度矩阵。(D)异构图推理
2 Materials and methods
药物定位的金标准数据集(Gottlieb等人,2011年)被用来证明我们所提出的方法的有效性。它包含1933个有效的药物-疾病协会,涉及593种药物和313种疾病。这些药物和疾病分别来自DrugBank(Wishart,2006)和在线孟德尔人遗传数据库(OMIM)(Ada等人,2002年)。对应的药物-疾病关联矩阵可以由二进制矩阵$A \in{0,1}^{\mathrm{n\times m}}$表示,其中m和n分别表示药物和疾病节点的数目。现有的药物-疾病对用1表示,而未知数用0表示。
对于药物,我们计算了5个药物相似性度量(Huang等,2020),包括化学结构相似性$R_{chem}$、解剖治疗性化学(ATC)编码相似性$R_{atc}$、副作用相似性$R_{se}$、药物-药物相互作用相似性$R_{ddi}$和靶向轮廓相似性$R_{targ}$。基于药物的经典Smiles(Weininger,1988)文件,我们使用化学开发工具包(CDK)(Steinbeck等人,2003)工具计算所有药物的散列指纹,然后获得$R_{chem}$。所有相关药品的ATC代码均从DrugBank中提取。我们应用了一种语义相似度算法(Resnik,1995)来计算ATC术语之间的相似度得分,从而得到$R_{atc}$。其余的相似性,包括$R_{se}$、$R_{ddi}$和$R_{targ}$,通过Jaccard相似系数(Jaccard,1908)来测量,该相似系数可以表示如下, $R_{se/ddi/targ}(i,j)=\frac{\left|S_{i} \cap S_{j}\right|}{\left|S_{i} \cup S_{i}\right|}$
其中$| \cdot |$表示集合的基数。SI分别代表药物I在RSE中的一组副作用图谱、药物I在RDDI中的药物-药物相互作用图谱和药物I在Rtarg中的药物-靶向相互作用图谱。具体地说,在RDDI中,每种药物都被表示为一个相互作用简档,由所有已知与其相互作用的药物组成。此外,从Sider(Kuhn等人,2016年)提取了药物的副作用,从DrugBank提取了药物-药物相互作用和药物-靶相互作用。